1. Introduction
In the field of cancer treatment, sulfoximine, which was found as a toxic compound in the 1940s, is now called Rising Star due to its unique significance in recent drug discovery [
1,
2,
3]. Since sulfoximines are the isosteres of sulfones with a mono-aza nitrogen atom [
4], they have special properties, such as the sulfur atom with an optical rotation property [
5,
6], the S=N bond with the double bond property, and nitrogen with weak nucleophilicity [
7]. Sulfoximines have been widely applied in asymmetric catalysis, synthesis of bioactive molecules, and modern pharmaceutical and agrichemical industries [
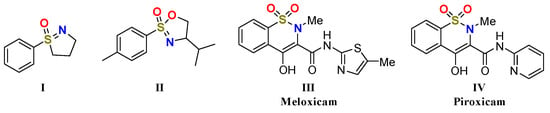
8]. Over the past decades, cyclic sulfoximines, as an important kind of sulfoximine derivatives, attracted a great deal of attention for chemists. Due to their outstanding biological properties, a number of molecules with potential medical application values appeared [
9], for example, five-membered cycle
I which performed well under in vitro pharmacokinetic studies [
10], and motif
II which could be transformed to potential scaffolds for peptide mimetics [
11]. Furthermore, analogues which also have the S=O bond and S–N bond, such as 1,2-benzothiazine 1,1-dioxides
III and
IV, were found to be anti-inflammatory drugs (
Figure 1) [
12].
Figure 1. Structures of bioactive cyclic sulfoximines and analogues.
It was once overlooked, but it has been re-emphasized recently that sulfoximine derivatives possess desirable properties as promising drug candidates. Additionally, C–H activation of sulfoximines has been widely considered to be an efficient method to construct complex scaffolds with potential bioactivities, especially since Bolm discovered the first efficient strategy of Rh-catalyzed annulation of sulfoximines and alkynes in 2013 [
13]. In addition, methyl phenyl sulfoximine (MPS) was proved to have better coordination adaptability to the transition metal center in C–H activation steps than some other nitrogen hetero groups [
14]. In a word, the cyclic sulfoximines exhibit great values, especially in the medicinal field.
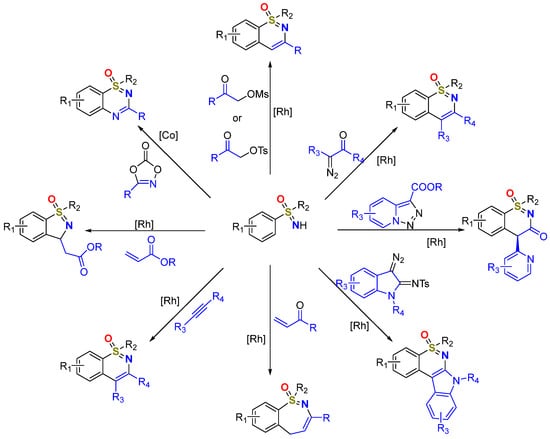
Additionally, the previously developed main approaches towards cyclic sulfoximines from sulfoximines are shown in
Figure 2. It was worth noting that the Co-catalyzed strategy was difficult to conduct smoothly but breakthroughs were also made in this area recently [
13,
20,
21,
22,
23,
24,
25,
26,
27].
Figure 2. The main strategies towards cyclic sulfoximines synthesis.
2. Synthesis of Cyclic Sulfoximines via Intermolecular C–H Activation
2.1. Metal Carbenoids as Coupling Partners
2.1.1. Diazo Compounds as Carbene Precursors
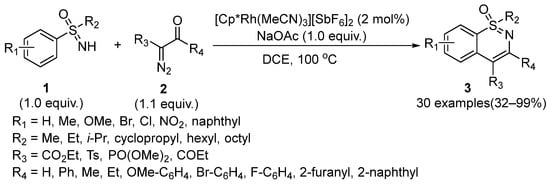
In 2015, Bolm and coworkers first described a Rh(III)-catalyzed intermolecular annulation between sulfoximines
1 and diazo compounds
2 for the synthesis of 1,2-benzothiazine products
3 (
Scheme 1) through the domino C–H activation/cyclization/condensation pathway [
21]. DCE was proved to be the optimal solvent and the yield of desired products reached up to 99% in the presence of NaOAc (1.0 equiv) as a base. Moreover, this kind of C–H activation featured wide substitution tolerance and afforded the desired adducts with low to excellent yields (32–99%).
Scheme 1. Rhodium-catalyzed annulation reactions for the synthesis of 1,2-benzothiazines.
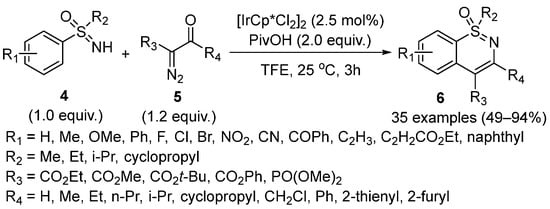
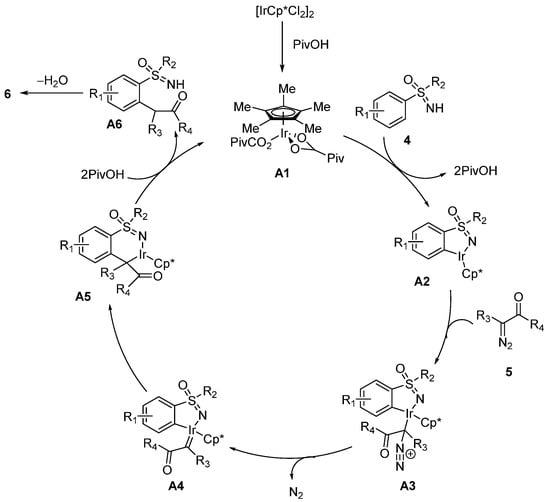
In 2018, the Ir(III)-catalyzed C–H functionalization of sulfoximines
4 with α-diazocarbonyl compounds
5 (
Scheme 2) was first developed by Pawar and coworkers [
28]. Furthermore, this C–H functionalization system was compatible with a broad scope of sulfoximines including electron-donating substituents, halogen substituents, and olefin motifs. However, the presence of strong electron-withdrawing groups such as NO
2 and CN significantly resulted in lower yields. Moreover, diazo compounds bearing various substituents were also tolerated in this protocol.
Scheme 2. Ir(III)-catalyzed C–H functionalization of sulfoximines with α-diazocarbonyl compounds.
The plausible mechanism (
Scheme 3) involves the following steps: firstly, neutral Ir(III) species
A1 with catalytic activity formed by [Cp*IrCl
2]
2 reacts with pivalic acid. Next, NH-sulfoximines
4 coordinate to the metal center to generate five-membered iridacycle
A2 via deprotonation and metallization. Subsequently,
A2 is coordinated with diazo precursors
5 and the loss of N
2 generates carbene species
A4. Then, the alkylated migratory insertion takes place, and subsequent protonolysis affords alkylated product
A6 and Ir(III) species
A1. Finally,
A6 undergoes the acid-promoted cyclization/dehydration process to give the expected product
6.
Scheme 3. Proposed mechanism of the formation of 1,2-benzothiazines in the Ir(III) catalytic system.
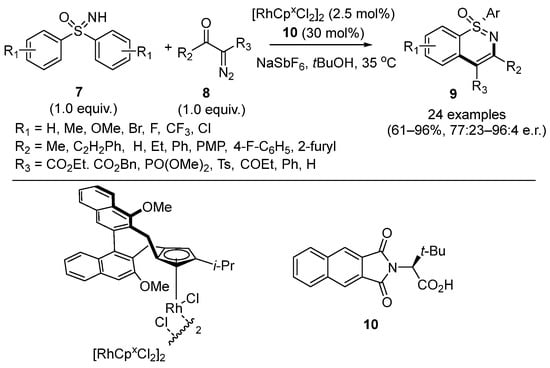
In the same year, Cramer’s group reported the enantioselective [4+2] annulative coupling reaction of diaryl sulfoximines
7 and acyl diazo compounds
8 for the synthesis of chiral-at-sulfur 1,2-benzothiazines
9 catalyzed by [RhCp*Cl
2]
2 and N-protected amino acid
10 (
Scheme 4) [
29]. Under the optimal condition, various substituted cyclic products were given with good to excellent yields (61–95%) and moderate to excellent enantioselectivities (77:23–96:4 e.r.). Moreover, it should be highlighted that the addition of polyfluoro-substituted alcohols such as TFE and HFIP as the solvents had a profound effect on the enantioselectivity for this reaction which gave the opposite main enantiomeric products.
Scheme 4. Synthesis of chiral-at-sulfur 1,2-benzothiazines via C–H functionalization of sulfoximines.
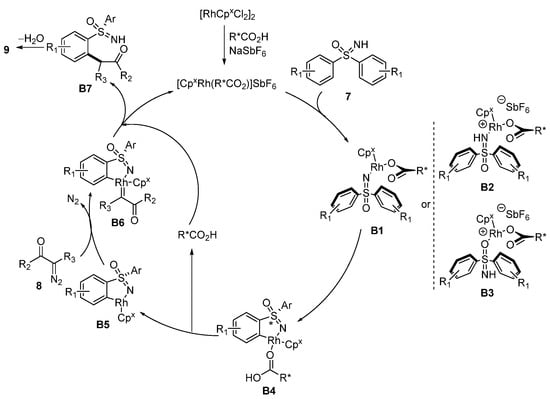
A plausible mechanism is shown in
Scheme 5, which is similar to the achiral version reported by Bolm. At first, sulfoximines
7 coordinate to the Rh(III) center to produce
B1 (possibly
B2 or
B3), and initiate the enantiodetermining
ortho-C–H activation step by a concerted metalation deprotonation pathway to give intermediates
B4. Then, five-membered cyclic rhodium complexes
B5 are formed by the auxiliary of the N-protected amino acid. Subsequently, carbenoid intermediates
B6, formed by coordination of the diazo species
8, undergo subsequent insertion and protonation to access
B7. Then, the product 1,2-benzothiazines
9 are generated by dehydration cyclization.
Scheme 5. Proposed mechanism of Rh-catalyzed chiral-at-sulfur 1,2-benzothiazines formation.
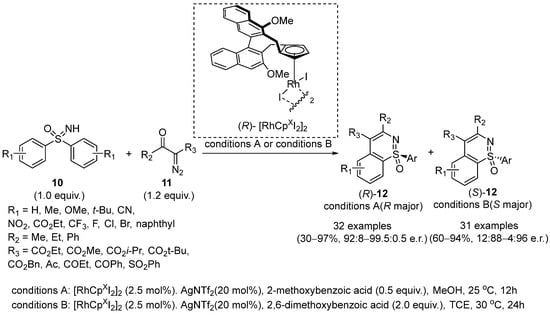
In 2018, Li and coworkers developed an enantiodivergent annulation between sulfoximines
10 and diazo compounds
11 catalyzed by rhodium(III) complexes [
30]. In this strategy, the authors employed the Cramer type Cp*Rh(III) complexes and various carboxylic acids with different steric bulks to invert the absolute configuration of the product, which gave (
R)-
12 and (
S)-
12, respectively, under the optimized conditions A and conditions B (
Scheme 6).
Scheme 6. Rhodium-catalyzed strategy for the enantiodivergent desymmetrization of sulfoximines.
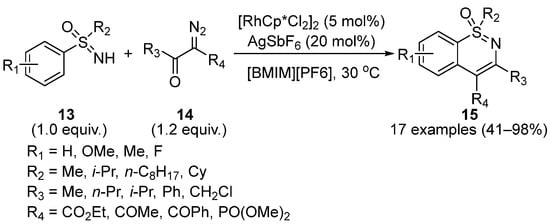
One year later, Wang and Wu reported a sustainable Rh(III)-catalyzed C–H activation/cyclization of sulfoximines
13 with diazo derivatives
14 to produce 1,2-benzothiazines
15 in the ionic liquid [BMIM][PF
6] (1-butyl-3-methylimidazolium-hexafluorophosphate) as a solvent (
Scheme 7) [
31]. This strategy exhibited a good tolerance for electron-donating and electron-withdrawing sulfoximine substituents. Moreover, this catalyst system could recycle at least 10 times before losing high-efficiency catalytic ability.
Scheme 7. Sustainable Rh(III)-catalyzed C–H activation/cyclization of sulfoximines to form 1,2-benzothiazines.
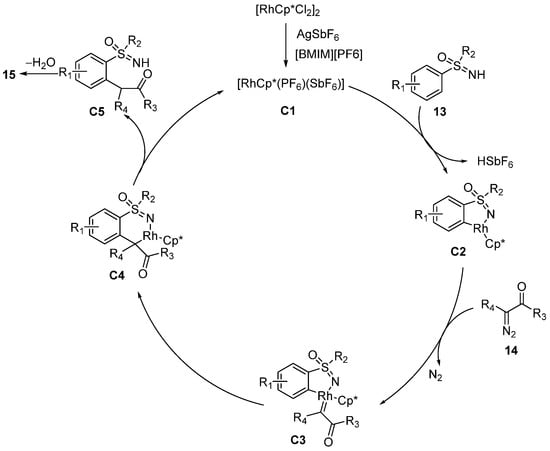
A proposed catalytic circle is shown in
Scheme 8. In the beginning, the dissociation of the ionic liquid [BMIM] [PF
6] gives PF
6* as a substitute anion to activate the catalyst [RhCp*Cl
2]
2 in the presence of AgSbF
6. Then, the activated catalyst
C1 is coordinated with sulfoximines
13 to form the five-membered Rh complexes
C2 and releases HSbF
6. Subsequently, diazo derivatives
14 coordinate to the Rh center and afford the carbene species
C3. Then, the insertion reaction of carbenes gives the six-membered intermediates
C4 and the following demetallization and intramolecular condensation furnish the product
15.
Scheme 8. Proposed catalytic circle.
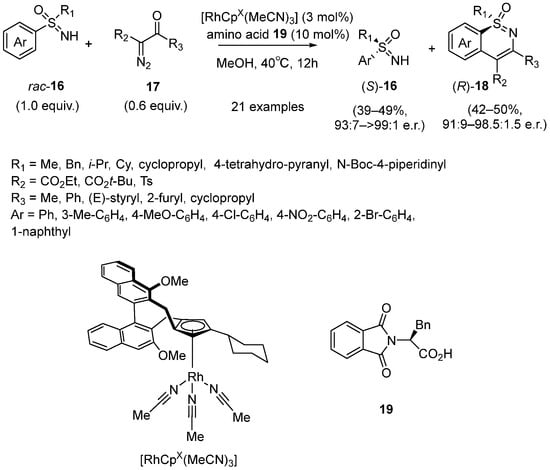
In the same year, Cramer developed a Rh(III)-catalyzed C–H functionalization strategy for the kinetic resolution of racemic sulfoximines
16 to generate S-chiral 1,2-benzothiazines
18 [32]. Compared to Cramer’s previous research in 2018, this approach could result in a higher e.r. value. In this case, sulfoximines were treated with diazoketoesters
17, giving the desired products in high
s-value (
Scheme 9). Moreover, different substituted aryl sulfoximines could react smoothly and the enantioselectivity was up to >99:1, including the groups with remarkable steric hindrance, such as
i-Pr and cyclohexyl.
Para-nitrophenyl methyl sulfoximine was a supreme substrate under these conditions, resulting in s-values of more than 200 with 47% yields for both of the two products.
Scheme 9. Rh(III)-catalyzed C–H functionalization strategy for the kinetic resolution of racemic sulfoximines.
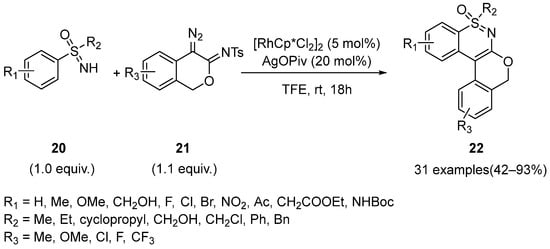
Highly fused polycyclic scaffolds exist widely in bioactive molecules, so their synthesis methods have also attracted much attention. In 2020, Li and Liu disclosed a Rh(III)-catalyzed method for the synthesis of fused 1,2-benzothiazines
22 from various sulfoximines
20 and 4-diazoisochroman-3-imines
21 (
Scheme 10) [
33]. Optimization studies showed that [RhCp*Cl
2]
2 (5 mol%) as a catalyst could work well with AgOPiv (20 mol%) as an additive under air in TFE at room temperature. In this strategy, haloalkanes were proved to be unfavorable to the yields because of the lack of polarity. Moreover, this reaction had a good substituent tolerance and resulted in moderate to excellent yields. However,
ortho- and
para-Me substrates could not form the desired products in high yields as expected, and the
meta-Br substrate could give a single site-selective product while the
meta-OMe substrate gave a mixture of two products.
Scheme 10. Rh(III)-catalyzed method for the synthesis of fused 1,2-benzothiazines from sulfoximines and 4-diazoisochroman-3-imines.
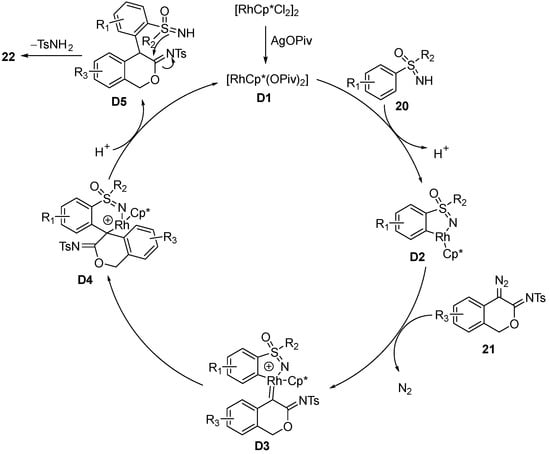
The mechanistic study demonstrated that the catalytic cycle (
Scheme 11) begins with the formation of the rhodacycle intermediate
D2, which is generated by the coordination of sulfoximine
20 to the Rh center of the activated catalyst [RhCp*(OPiv)
2]
D1 and the electrophilic C–H bond cleavage. Next, the rhodium carbenoid complex
D3 is formed by the coordination of
D2 with diazo compound
21, via the loss of nitrogen gas. Then, the insertion to the Ph–Rh bond gives intermediate
D4, and subsequently, the catalyst is recycled by protonation of
D4, giving intermediate
D5. At last, the product
22 is obtained by the condensation of
D5 with the loss of TsNH
2.
Scheme 11. Proposed mechanism of the annulation between sulfoximines and 4-diazoisochroman-3-imines.
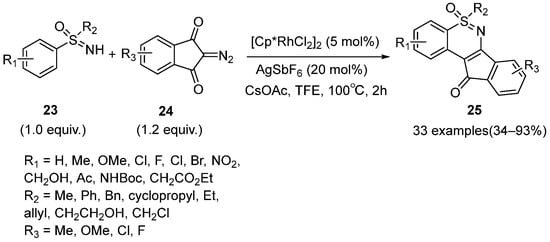
In 2021, Ye and Liu and coworkers described a methodology for the synthesis of highly conjugated 1,2-benzothiazine scaffolds
25 using sulfoximines
23 and 2-diazo-1H-indene-1,3(2H)-diones
24 (
Scheme 12) [
34]. The silver additive was proved to be important for high yields. Moreover, this protocol tolerated a wide range of substituted sulfoximines and provided target products with 74%–88% yields. Furthermore, it was worth mentioning that the C–H bond activation of
meta-substituted sulfoximines tended to proceed on the more hindered
ortho-positions. Various mono-substituted diazo compounds could also react smoothly, but the corresponding products were a regional isomers mixture.
Scheme 12. Rh-catalyzed synthesis of highly conjugated 1,2-benzothiazine scaffolds.
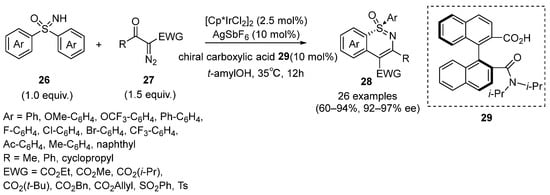
Very recently, Hong and Shi reported an Ir(III)-catalyzed C–H activation/annulation strategy for the synthesis of chiral 1,2-benzothiazines
28 by the desymmetrization of sulfoximines
26 with diazo compounds
27 in the presence of chiral carboxylic acid
29 as ligand (
Scheme 13) [
35]. Various electron-withdrawing groups on the diazo compounds were screened to be well tolerated and the ee value was up to 97%. Both electron-donating and electron-withdrawing substituents of sulfoximines could also transform smoothly. Notably, the kinetic resolution and parallel kinetic resolution of racemic sulfoximines with this protocol were also developed by the authors.
Scheme 13. Ir(III)-catalyzed C–H activation/annulation strategy for the synthesis of chiral 1,2-benzothiazines.
2.1.2. Ylide Active Intermediates as Carbene Precursors
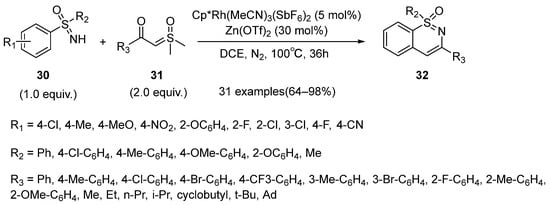
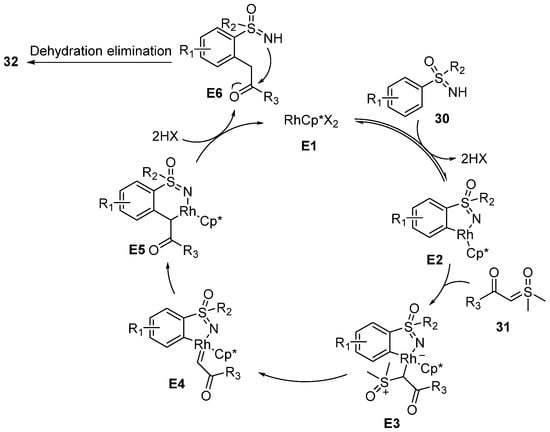
In 2018, Li’s group reported a novel rhodium(III)-catalyzed annulative coupling strategy for the synthesis of 1,2-benzothiazines
32 between sulfoximines
30 and sulfoxonium ylides
31 (
Scheme 14) [
36]. Various substituted substrates were compatible. Moreover, different N-protected benz-amidines and benzophenone NH-imines were also tolerated, and they could provide various isoquinoline products in good yields. In order to clarify the reaction mechanism, a H/D exchange experiment was conducted and the results showed that partial H/D exchange was appeared at the
ortho- position of the product. It was obvious evidence to reveal that the C–H activation is reversible. Moreover, the kinetic isotope effect of this reaction was also measured, and the result suggested that the C–H activation step was involved in the turnover-limiting process.
Scheme 14. The synthesis of 1,2-benzothiazine through rhodium(III)-catalyzed annulative couplings.
The proposed catalytic mechanism is presented in
Scheme 15. At first, the activated RhCp*X
2 complex
E1 is coordinated with sulfoximine
30 to form intermediate
E2 by a metalation-deprotonation process. Then, the sulfoxonium ylides
31 coordinate to the rhodium center to generate intermediate
E3. Subsequently, α-elimination of DMSO occurs, which leads to an active carbene complex
E4. In addition, the Rh–Ar bond of the carbene species undergoes the migratory insertion pathway to generate intermediate
E5. At last, reductive elimination and intramolecular dehydration obtain product
32.
Scheme 15. Proposed mechanism of the tandem annulative coupling between sulfoximines and sulfoxonium ylides.
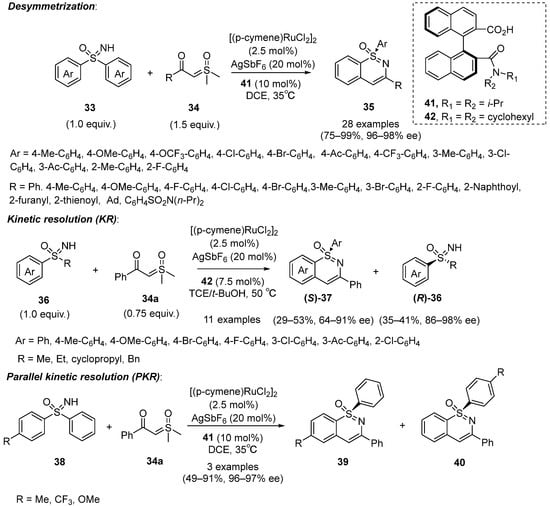
In 2021, Shi’s group reported an enantioselective C–H functionalization of sulfoximines
33 with sulfoxonium ylides
34 using chiral binaphthyl carboxylic acids
41 as the optimal chiral ligand in combination with [(p-cymene)RuCl
2]
2 as a catalyst to obtain 1,2-benzothiazines
35 with good to excellent yields (75–99%) and excellent enantioselectivities (96–98% ee) (
Scheme 16) [
37]. Moreover, the kinetic resolution of racemic sulfoximines
36 could also be achieved when chiral binaphthyl carboxylic acids
42 were selected as the ligand, which produced the cyclization products (
S)-
37 with low to moderate yields (29–53%) and moderate to excellent enantioselectivities (64–91%). Furthermore, the unreacted (
R)-
36, with low to moderate yields (35–41%) and excellent enantioselectivities (86–98%), could also be obtained. Fortunately, parallel kinetic resolution of racemic diaryl sulfoximine
38 could be carried out smoothly with high ee values (up to 95%) under the standard conditions.
Scheme 16. Desymmetrization, kinetic resolution, and parallel kinetic resolution of sulfoximines with sulfoxonium ylides under the Ru-catalyzed system.
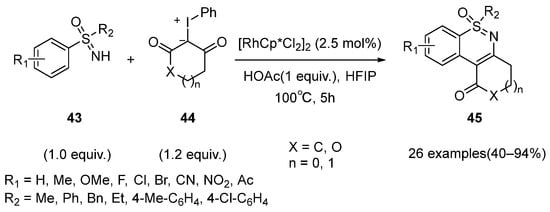
In 2021, Yu and Pan disclosed a rhodium-catalyzed strategy for the synthesis of polycyclic 1,2-benzothiazines
45 using different aryl sulfoximines
43 and iodonium ylides
44 as substrates (
Scheme 17) [
38]. Through the catalyst screening, [RhCp*Cl
2]
2 was proved to be the most efficient catalyst for this reaction system. Moreover, various polycyclic 1,2-benzothiazines with good functional group tolerance were furnished in moderate to excellent yields.
Scheme 17. Rhodium-catalyzed strategy for the synthesis of polycyclic 1,2-benzothiazines from different aryl sulfoximines and iodonium ylides.
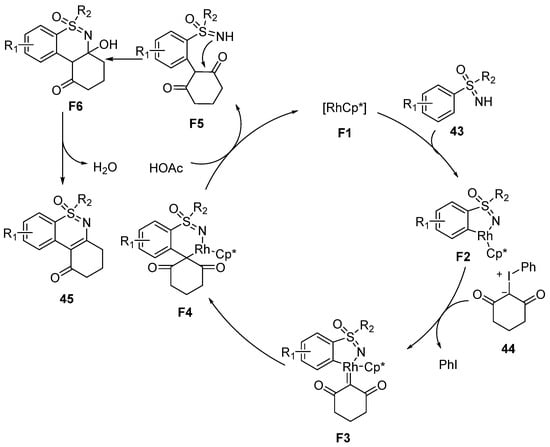
In order to reveal the reaction mechanism, the H/D exchange experiment and KIE studies were conducted. The results indicated that the C–H activation step may be irreversible and the cleavage of the C–H bond may not be involved in the rate-determining step. Moreover, the intermolecular competition experiments were also conducted and the results confirmed that electron-donating sulfoximines were beneficial to the C–H functionalization process. The proposed mechanism was similar to the previous reports (
Scheme 18). At first, sulfoximines
43 coordinate to the Rh-catalysis
F1 to achieve C–H activation and yield a five-membered cyclic complex
F2. Then,
44 reacts with
F2 to generate a carbene species
F3 along with the loss of PhI. Subsequently, the migratory insertion of Rh–carbene into the C–Rh bond is conducted, and obtains intermediate
F4. After the reductive elimination of
F4, the intramolecular nucleophilic cyclization and dehydration take place to form the desired product
45.
Scheme 18. Proposed mechanism of the reaction between sulfoximines and iodonium ylides.
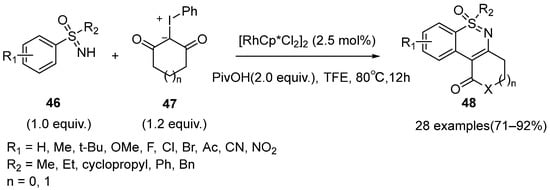
Almost simultaneously, Wu et al. reported a very similar approach to provide polycyclic 1,2-benzothiazines
48 in good to excellent yields (
Scheme 19). In this case, trimethylacetic acid (PivOH) was added as an additive [
39]. Similar with Yu’s work, this protocol exhibited a broad substrate scope and was easy to obtain cyclohexanone-1,2-benzothiazine scaffolds.
Scheme 19. Rhodium-catalyzed strategy for the synthesis of polycyclic 1,2-benzothiazines.
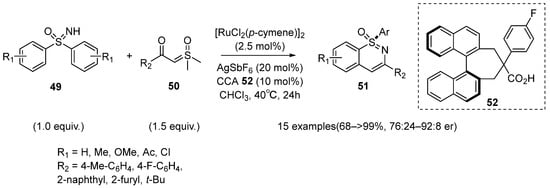
In 2022, Yoshino et al. developed a Ru(II)-catalyzed C–H activation/annulation of sulfoximines
49 with sulfoxonium ylides
50 for the enantioselective synthesis of 1,2-benzothiazines
51 (
Scheme 20) [
40]. The pseudo-C2-symmetric binaphthyl monocarboxylic acid
52 (10 mol%) proved to be the most optimal chiral ligand. A broad range of S-chiral 1,2-benzothiazine products were prepared in moderate to excellent yields with up to 92:8 er. Moreover, this protocol showed a compatibility for electron-donating substituents, electron-withdrawing substituents, and steric hindrance substituents.
Scheme 20. Ru(II)-catalyzed C–H functionalization strategy of sulfoximines for the enantioselective synthesis of 1,2-benzothiazines.
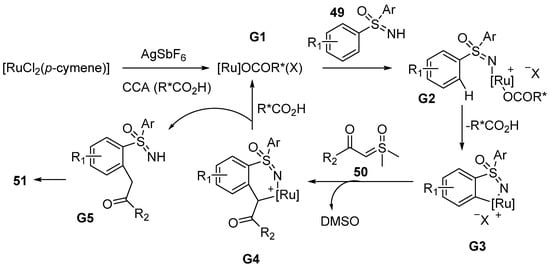
A plausible catalytic cycle is outlined in
Scheme 21. It includes five main steps. At the beginning, a Ru–carboxylate catalyst complex
G1 is formed by [RuCl
2 (p-cymene)]
2, AgSbF
6, and
52. Next, the sulfoximines
49 coordinate to
G1, to assist C–H activation via a selective deprotonation process to give intermediate
G3. Then, the chiral metallacycle complex
G3 coordinates with sulfoxonium ylides
50, and next, the insertion of carbene to the Ru–Aryl bond gives a six-membered cycle
G4, with the loss of DMSO in the reaction process. At last, the protonation and intramolecular condensation take place to access the desired products
51.
Scheme 21. Proposed mechanism of Ru-catalyzed enantioselective synthesis of 1,2-benzothiazines.
2.2. Metal Nitrene as Coupling Partner
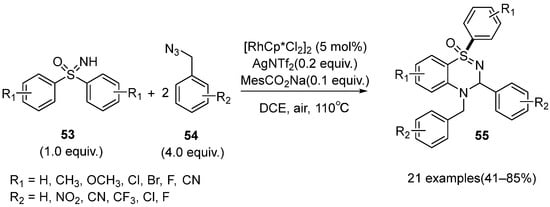
In 2019, Rh(III)-catalyzed C–H activation/cyclization for the synthesis of dihydrobenzo thiadiazine 1-oxide derivatives
55 using sulfoximines
53 and benzyl azides
54 as starting materials was reported by Xu and Dong (
Scheme 22) [
41]. This catalytic reaction was tolerant to various substrates bearing various electron-donating and electron-withdrawing groups, but the heterocyclic NH-sulfoximines and fluorinated or free benzyl azides afforded poor results. Additionally, the cyclization products were obtained in moderate to good yields (41–85%).
Scheme 22. Rh(III)-catalyzed C–H functionalization of sulfoximines to prepare dihydrobenzo thiadiazine 1-oxide derivatives.
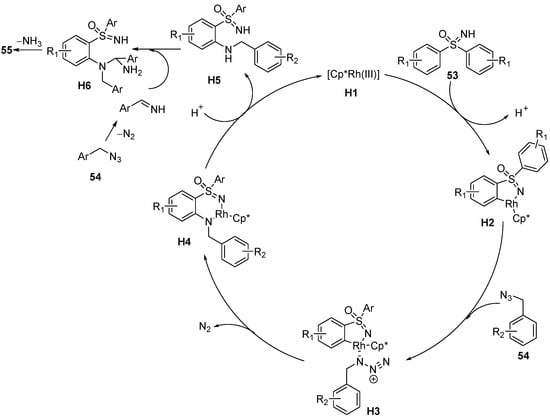
Based on a series of control experiments, a proposed mechanism is suggested in
Scheme 23. Firstly, the sulfoximines
53 coordinate to the activated Rh(III) compound
H1 and subsequently undergo the C−H bond cleavage process to generate five-membered rhodacyclic complexes
H2. After the coordination of the benzyl azides
54, the nitrene intermediates
H3 are formed. Then, the migratory insertion occurs, and the C−N bond formation leads to a six-membered rhodium cycle
H4 with the loss of N
2. Finally, protonolysis of
H4 leads to the formation of
H5 and recycle catalyst
H1. Further cyclization leads to the formation of the observed products
55 with the loss of NH
3.
Scheme 23. Proposed mechanism of C–H functionalization of sulfoximines and annulation with benzyl azides.
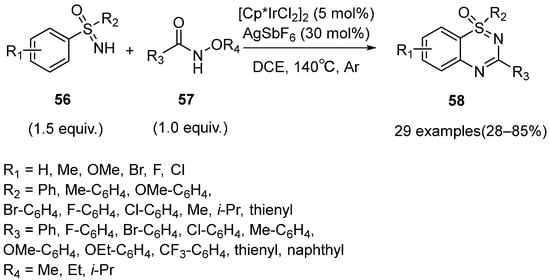
One year later, Dong’s group disclosed an Ir(III)-catalyzed amidation/cyclization of sulfoximines
56 with N-alkoxyamides
57 to synthesize thiadiazine 1-oxides
58 in low to high yields (28–85%) (
Scheme 24) [
42]. This method featured diverse substituents and functional groups tolerance, but the substrates with strong electron-withdrawing groups did not react smoothly.
Scheme 24. Ir(III)-catalyzed amidation/cyclization for the C–H activation of sulfoximines with N-alkoxyamides to synthesize the corresponding thiadiazine 1-oxides.
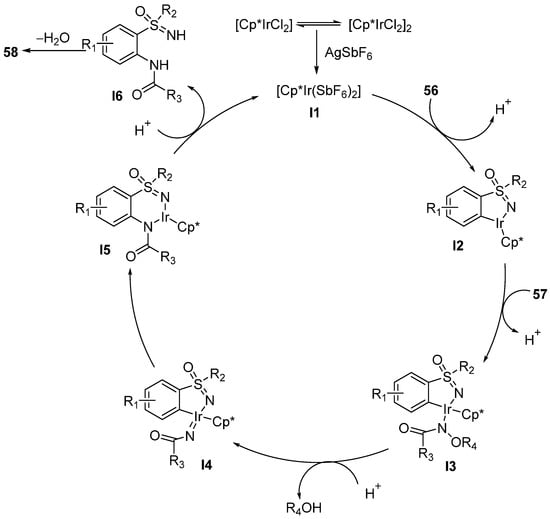
According to the reaction mechanism (
Scheme 25), the cleavage of the N–O bond of the intermediate
I3 was considered to be a key step, which generates the nitrene complex
I4 by protonation.
Scheme 25. Proposed catalytic cycle.
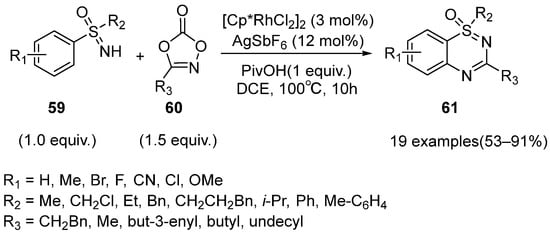
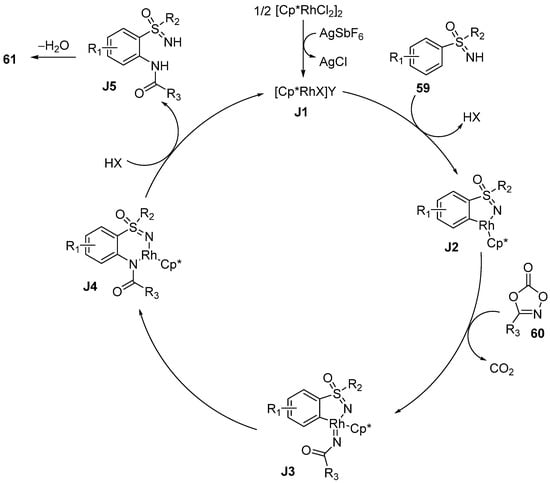
In 2020, Bolm and coworkers reported an effective Rh(III)-catalyzed C–H functionalization cyclization strategy to prepare benzothiadiazine-1-oxides
61 from various sulfoximines
59 and 1,4,2-dioxazol-5-ones
60 (
Scheme 26) [
43]. This protocol could be conducted in the air without other protecting strategies.
Scheme 26. Rh(III)-catalyzed method to obtain benzothiadiazine-1-oxides using 1,4,2-dioxazol-5-ones.
The results of the substrate scope studies show that all of the substrates with electron-donating, electron-withdrawing groups, and huge steric hindrance afforded the benzothiadiazine-1-oxides 61 in moderate to excellent yields (53–91%). Moreover, this work was well-suited for S-alkyl-S-aryl-substituted sulfoximines, which were specific comparing with the previous reports.
Consistent with the previously reported mechanism (
Scheme 27), the five-membered rhodacycles
J2 are formed by the reaction of sulfoximines
59 and the activated Rh(III) catalyst. Then, they react with
60 to generate nitrenoid intermediates
J3, accompanied by the loss of CO
2. Next, the products
61 are obtained via a classical insertion–demetallization–condensation pathway.
Scheme 27. Proposed mechanism.
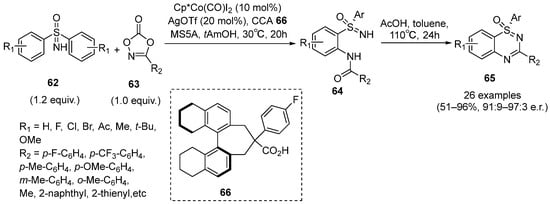
In 2022, Yoshino and coworker described a protocol involving Cp*Co(CO)I
2/chiral carboxylic acid
66 as a catalytic system, which promoted the C–H functionalization reaction of sulfoximines
62 with dioxazolone
63 for the enantioselective synthesis of benzothiadiazine-1-oxides
65 (
Scheme 28) [
44]. This strategy consisted of two reaction steps. Firstly, under the transition metal catalyst system, sulfoximines
62 reacted with dioxazolones
63 to form the amidation product
64. Then,
64 was treated with AcOH in toluene to give the cyclic products with moderate to excellent yields and good to excellent e.r. values. Moreover, the control experiments and DFT calculation were performed to confirm that the cleavage of the C–H bond is the enantiodetermining step. Furthermore, the studies of the scope of the substrates showed that this protocol has a good compatibility with several functional groups.
Scheme 28. Co(III)-catalyzed enantioselective C–N bond formation of cyclic sulfoximines.
2.3. Alkenes as Coupling Partner
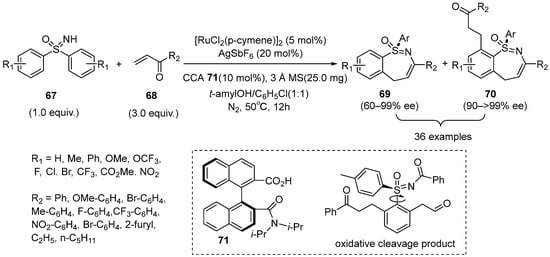
In 2022, Shi’s group developed an asymmetric [4+3] annulation between sulfoximines
67 and α,β-unsaturated ketones
68, which was promoted by the Ruthenium(II) /chiral carboxylic acid
71 catalyst system. It generated a broad range of sulfur-stereogenic 1,2-benzothiazepine 1-oxides with up to 90% yield and up to >99% ee. It is worth noting that the positions of electron-withdrawing and electron-donating groups often led to the different main products. For example,
para-substituted diarylsulfoximines bearing electron-donating groups (OMe, Ph) often benefited to the dialkylation/cyclization products, and the
para- or
meta-substituted diarylsulfoximines sulfoximines with electron-withdrawing groups would help to access mono alkylation/cyclization products. Moreover, a series of chiral N-benzoyl sulfoximines with a C–S chiral axis could be obtained after the oxidative cleavage of the double bonds in the products (
Scheme 29) [
45].
Scheme 29. Ru(II)/chiral-carboxylic-acid-catalyzed enantioselectivity synthesis of seven-membered cyclic sulfoximines.
2.4. Alkynes as Coupling Partner
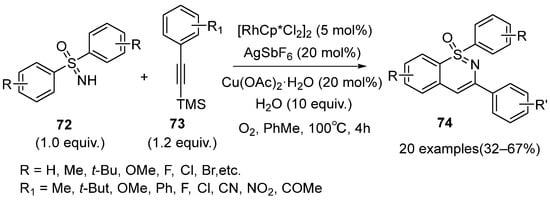
It was generally considered that terminal alkynes were incompatible with the Rh(Ⅲ) catalytic system because of the existence of the active hydrogen atoms. In 2019, Prabhu et al. reported a novel strategy for the synthesis of 1,2-benzothiazines
74 using diphenyl sulfoximines
72 and alkynyl silanes
73 as substrates catalyzed by the Rh(Ⅲ) catalytic system (
Scheme 30) [
46]. In the presence of AgSbF
6 and H
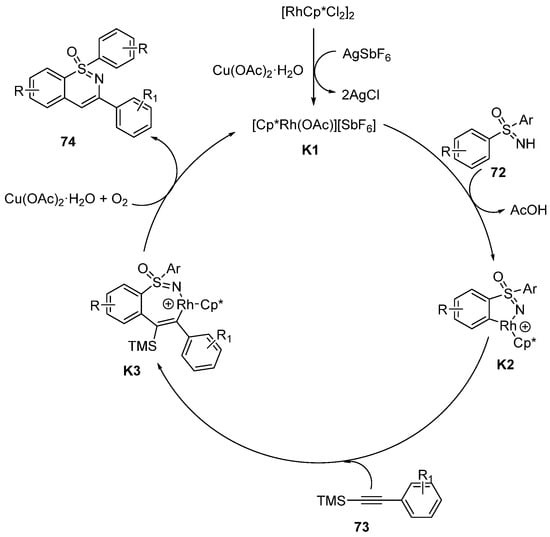
2O, the TMS group was facilely removed from the alkynyl silanes to give the desired motifs. Oxygen was necessary for this reaction. A series of 1,2-benzothiazine derivatives with a free substituent at the C-4 position were obtained in low to moderate yields (32–67%), which had the potential to transform into other functional scaffolds. In addition, the degradation of the sulfoximines may be the reason for the relatively low yields.
Scheme 30. The synthesis of 1, 2-benzothiazines in the Rh(Ⅲ) catalytic system using alkynyl silanes.
The plausible mechanism is demonstrated in
Scheme 31. In the beginning of the cycle, AgSbF
6 and Cu(OAc)
2·H
2O play a key role for the activation of the catalyst. Next,
K1 coordinates with sulfoximines
72 to form the five-membered rhodacycle complexes
K2. Then, the insertion of arylalkynyl silanes
73 provides the intermediates
K3. At last, the products
74 are obtained by reductive elimination of
K3, and the active catalyst
K1 is also regenerated.
Scheme 31. Proposed mechanism of annulation of sulfoximines and alkynyl silanes in the Rh catalytic system.
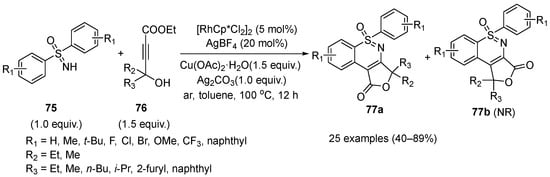
In the same year, Prabhu developed a Rh(Ⅲ)-catalyzed cascade C−H activation, regioselective annulation, and lactonization strategy for the synthesis of furanone-fused 1,2-benzothiazines derivatives
77a using sulfoximines
75 and 4-hydroxy-2-alkynoates
76 (
Scheme 32) [
47]. The reaction exhibited good functional group tolerance and displayed regioselectivity in forming single regioisomers
77a in moderate to good yields. The other regioisomer
77b was not detected, which may be due to the steric interaction between the hydroxyl oxygen atom and the Rh(III) center.
Scheme 32. Tandem Rh(Ⅲ)-catalyzed C–H activation of sulfoximines to furnish furanone-fused 1,2-benzothiazines derivatives.
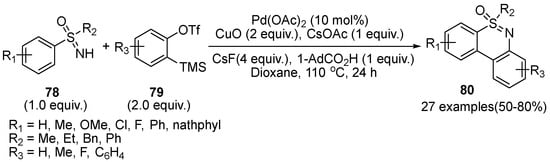
In 2020, Li et al. demonstrated Pd(II)-catalyzed C–H functionalization/cyclization of sulfoximines
78 with aryne precursors
79 to prepare tricyclic dibenzothiazines
80 by utilizing Pd(OAc)
2 as the optimal catalyst, and CuO as the oxidant (
Scheme 33) [
48]. A variety of sulfoximines bearing electron-rich, halogen, and S-substituents performed well and the naphthyl- sulfoximine afforded a mixture of regioselective products.
Scheme 33. Pd(II)-catalyzed C–H functionalization strategy for the preparation of dibenzothiazines.
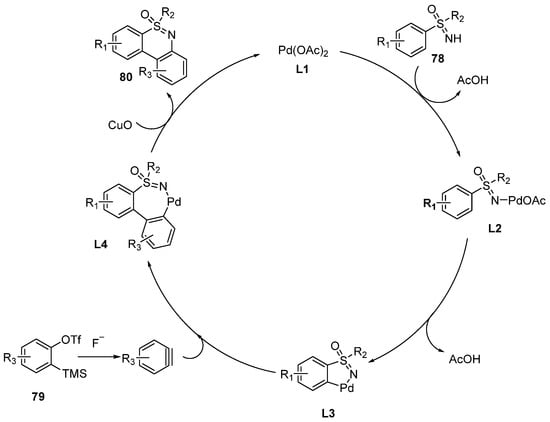
The proposed mechanism is shown in
Scheme 34. Initially, Pd(OAc)
2 reacts with sulfoximines
78 directly to generate the acyclic Pd species
L2 accompanied by the release of AcOH. Then, five-membered palladium cyclic complexes
L3 are formed by the intramolecular deprotonation with the loss of another molecule of AcOH. Subsequently, benzyne insertion occurs and produces seven-membered cyclic palladium complexes
L4. At last,
L4 undergo a reductive elimination pathway to furnish the products
80. Simultaneously, the activated catalyst
L1 is regenerated by reoxidation of Pd(0) to Pd(II) in the presence of the copper salt.
Scheme 34. Proposed mechanism of Pd-catalyzed annulation of sulfoximines with arynes.
3. Synthesis of Cyclic Sulfoximines via Intramolecular C–H Activation
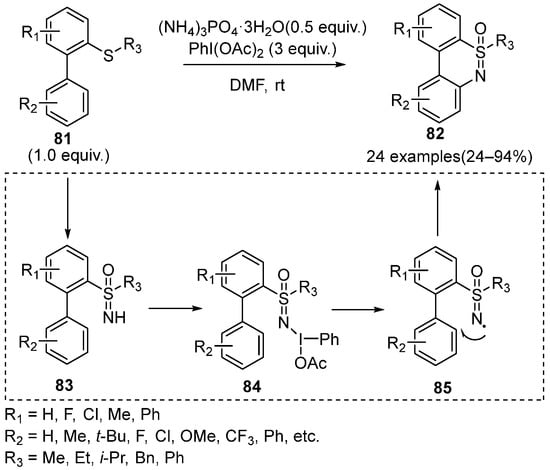
In 2018, Zhang et al. reported a metal-free method to synthesize benzothiazines
82 through one-pot N,O-transfer and intramolecular C–H amination of 2-biphenylsulfoximines
81 (
Scheme 35) [
49]. The results of reaction conditions screening showed that selecting (NH
4)
3PO
4·3H
2O as the N-source and PhI(OAc)
2 as the oxidant was necessary to generate the products
82 in high yields. Noteworthy, the mechanism studies proposed that PhI(OAc)
2 could perform as a free radical initiator for NH-sulfoximines to form the crucial intermediate
84 and
85 in the plausible pathway. Moreover, this protocol has a wider universality to the substrates. Various dibenzothiazine derivatives could be conveniently accessed in the yield ranging from 24–94%.
Scheme 35. Metal-free method for the intramolecular C–N coupling to form benzothiazines.
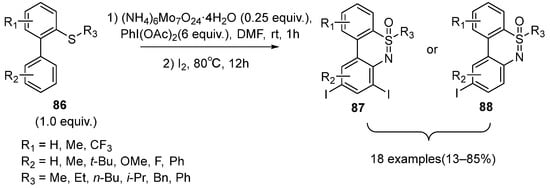
Based on the previous studies, Zhang’s group further investigated the one-pot N,O-transfer and intramolecular C–H amination of 2-biaryl sulfides
86 to afford a series of iodo-dibenzothiazines compounds
87 (
Scheme 36) [
50]. However, monoiodo-dibenzothiazines
88 were also observed when the 4′-positions of sulfides were substituted by large steric hindrance substituents or the strong electron-withdrawing substituents such as CF
3. Moreover, the desired products could be further transformed to other functional thiazine compounds through the cross-coupling reaction.
Scheme 36. Tandem C–H activation and C–N coupling of sulfoximines to form iodo-thiazines.
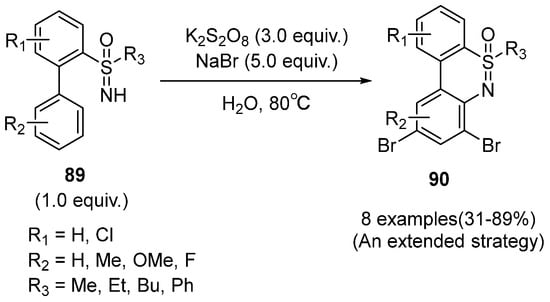
In 2019, Ma and coworkers reported a metal-free tandem amination/bromination method for the synthesis of bromobenzothiazines
90 by intramolecular C–H activation of N-methyl-2-biphenylsulfonamides and 2-biphenylsulfoximines
89 (
Scheme 37) [
51]. Interestingly, different from the sulfonamides substrates, sulfoximines could afford dibromothiazines as the only products and the momo-bromothiazines were not detected under the standard conditions.
Scheme 37. Metal-free tandem amination/bromination method for the synthesis of bromobenzothiazines.
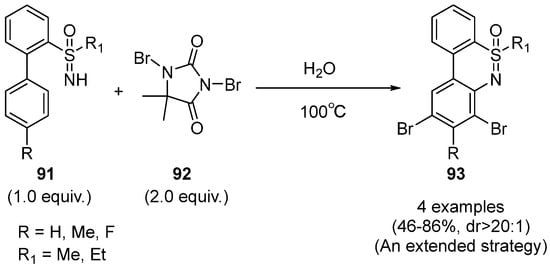
One year later, Zhang and Chen developed a step-economic DBH-promoted cyclization strategy for the synthesis of bromo-N-heterocycles
93 from the 2-biphenyl sulfoximines
91 in water (
Scheme 38) [
52]. Moreover, this novel strategy fulfilled the synthesis of various N-heterocycles products in moderate to good yields and up to >20:1 dr, and the substrate scope was also extended to 2-biphenyl phosphamides.
Scheme 38. DBH-promoted strategy for the cyclization of sulfoximines.
4. Miscellaneous
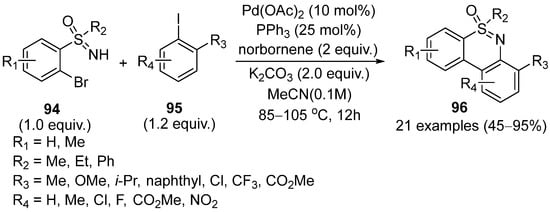
In 2018, Chen and coworkers reported the Pd/norbornene-catalyzed coupling reaction for the synthesis of fused polycyclic sulfoximines
96 with 2-bromo-NH-sulfoximines
94 and aryl iodides
95 (
Scheme 39) [
53]. PPh
3 was confirmed to be the optimal ligand and norbornene was proved to be necessary for this reaction. The scope of substrates was also explored under the optimized conditions. Reaction of aryl iodides and ortho-bromophenyl sulfoximines successfully afforded the desired products in good yields. Moreover, aryl iodides with electron-withdrawing groups (CO
2Me and CF
3) also transformed smoothly. However,
ortho-substituent would affect the reaction process.
Scheme 39. Pd/norbornene-catalyzed coupling reaction for the synthesis of fused polycyclic sulfoximines.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28031367