Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Rheumatology
Glucocorticoid-induced osteogenic dysfunction is the main pathologyical mechanism underlying the development of glucocorticoid-induced osteoporosis. Glucocorticoids promote adipogenic differentiation and osteoblast apoptosis through various pathways. Various ongoing studies are exploring the potential of natural products in preventing glucocorticoid-induced osteoporosis. Preclinical studies have consistently shown the bone protective effects of tocotrienol through its antioxidant and anabolic effects.
- antioxidant
- anti-inflammatory
- bone
- osteoblasts
- osteoporosis
1. Introduction
Osteoblasts are specialised bone-forming cells that play many roles in bone remodelling. Osteoblasts develop from multipotent mesenchymal cells under the influence of regulatory transcription factors runt-related transcription factor 2 (RUNX2) and osterix, which also can grow into other cell lineages such as adipocytes, myocytes and chondrocytes [1]. Osteoblasts produce organic bone matrix (osteoid) and subsequently mineralise it during skeletal modelling and bone remodelling [2]. The differentiation and function of osteoblasts are regulated by several signalling pathways, one of which is the canonical Wingless and Int-1 (Wnt) signalling pathway. It is activated by the binding of Wnt1 and Wnt3a protein with Fizzled and low-density lipoprotein receptor-related protein 5/6. This leads to the inhibition of glycogen synthase kinase-3 beta, which phosphorylates cytoplasmic beta-catenin, causing its degradation. The accumulation of beta-catenin leads to its nuclear translocation, forming a complex with T-cell factor/lymphocyte enhancer factor 1 and cAMP response element-binding-binding protein and translation of genes responsible for osteoblast differentiation and bone formation. Sclerostin (SOST) and dickkopf-1 (DKK1) are inhibitors of the Wnt signalling pathway secreted by osteocytes [3,4]. Osteoblasts also secrete receptor activator of nuclear factor kappa-Β (RANK) ligand (RANKL), which binds with RANK on osteoclasts precursors and stimulates their differentiation. At the same time, osteoblasts also secrete osteoprotegerin (OPG), which is a decoy receptor for RANKL, to prevent the binding of RANKL with RANK and suppress osteoclast formation. These signalling pathways are influenced by various endogenous and exogenous factors, including inflammatory cytokines, oxidative stress and glucocorticoids (GCs) [5].
GCs are widely used as the treatment for inflammatory diseases and as chemotherapeutic agents. They are crucial for the induction of osteoblast differentiation and the formation of a mineralised extracellular matrix, as they influence the signals coordinating the differentiation of multipotent mesenchymal precursors cells into osteoblasts [6,7,8]. GCs have been reported to stimulate the expression of differentiation markers of both osteoblasts and adipocytes in in vitro studies [9]. However, the prolonged use of GCs leads to osteoporosis. High GC levels have been reported to suppress osteoblastic differentiation and function [10] and induce osteoblast apoptosis, leading to the suppression of bone formation [11]. The effects of high GC on osteoblast are mainly mediated by suppression of the Wnt signalling pathway [12].
The current prevention for GC-induced osteoporosis (GIO) is ensuring an adequate intake of vitamin D and calcium among patients on GCs. The assessment of fall risk in patients taking high doses of GCs should be performed. Lifestyle modifications similar to those adopted by patients with postmenopausal osteoporosis, which include weight-bearing exercise and the elimination of other risk factors, such as smoking or alcohol, should also be implemented by chronic GC users [13]. Patients using GCs at moderate or high osteoporosis risk are recommended to start with pharmacologic agents. The first-line agent is bisphosphonate therapy due to its efficacy, safety and low cost. Bisphosphonates, such as once-weekly oral alendronate or once-monthly oral ibandronate, have been the most studied agents for the prevention and treatment of GIO. Treatment with bisphosphonates has been proven to improve bone mineral density (BMD) or vertebral fracture risk and reduced the hip fracture risk [14]. If bisphosphonates are contraindicated, teriparatide is suggested as the second-line agent, which is followed by denosumab. Teriparatide, an anabolic agent, has also been shown to reduce fracture risk [15]. Denosumab, a receptor activator of nuclear factor kappa-Β (RANK) ligand (RANKL) inhibitor, has been reported to increase lumbar spine and hip BMD. However, the data regarding its efficacy and safety are still very limited [16]. The search for other alternative agents to preserve skeletal health in GC users is still ongoing.
Vitamin E encompasses both tocotrienol and tocopherol, both of which are potent antioxidants. They could be divided into alpha (α), beta (β), gamma (γ) and delta (δ) isomers. Tocotrienol and tocopherol share a similar structure, which consists of a chromanol ring and a long carbon tail. Tocotrienols have three double bonds, whereas tocopherols have only single bonds on the carbon tail (Figure 1) [17]. The main sources of tocotrienol include rice bran, palm, and annatto. Palm-derived tocotrienol (PTT) contains approximately 75% tocotrienol and 25% tocopherol, whereas annatto-derived tocotrienol (ATT) contains solely tocotrienol (99.9%) [18]. Previous studies have established the role of tocotrienol as an anti-oxidative, anti-inflammatory, anti-hyperlipidaemic and anti-hyperglycaemic agent [18,19,20,21].
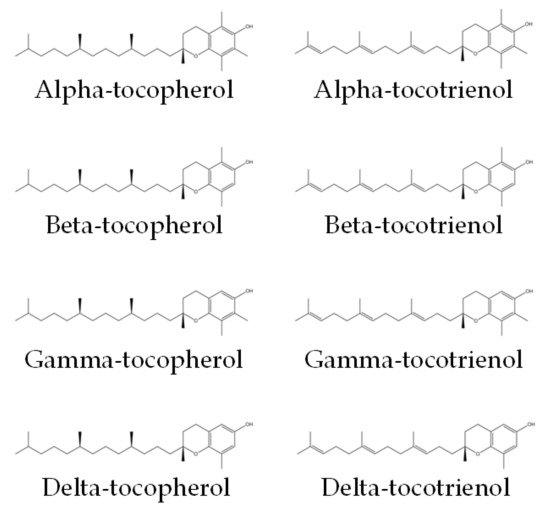
Figure 1. Chemical structures of tocopherols and tocotrienols [22]. They consist of a chromanol ring and a long carbon tail. On the carbon tail, tocotrienols have three double bonds, whereas tocopherols have only single bonds.
2. Glucocorticoids and Bone
2.1. Direct Action of Glucocorticoid on Osteoblasts
GC-induced osteoblast dysfunction is one of the main pathological mechanisms underlying the development of GIO [23]. GCs impair osteoblast proliferation, increase apoptosis and alter autophagy through changing RANKL/OPG and Wnt/sclerostin expression. Their inhibitors, microRNAs, IL-11, BMP/notch signalling, and effectors of apoptosis also play a major role in the action of GCs on the osteoblasts [24]. GCs suppress the canonical Wnt signal pathway that stimulates osteoblast differentiation and activity, partially through the enhancement of the DKK1 production in cultured human osteoblasts [25]. Using dexamethasone (DEX) as an example, at higher concentrations, it inhibits osteoblast differentiation by decreasing alkaline phosphatase (ALP) activity, RUNX2 and osteocalcin (OCN) expressions, and increasing RANK expression [25,26].
The actions of GCs on bone are also determined by variations in the expression and sensitivity of the GC receptors, the export of steroids from the cell by transmembrane transporters, and enzymatic metabolism of GCs to the more or less active metabolites [27]. 11β-hydroxysteroid dehydrogenases (11β-HSDs), which control the interconversion between the active cortisol and corticosterone and their inactive counterparts, cortisone and dehydrocorticosterone, also contribute to the skeletal action of GC on bone [27]. All these events contribute to increased bone turnover, reduced mineralization, and subsequently, bone loss.
GCs are important in the differentiation of osteoblasts, but their effects on osteoblast proliferation are inconsistent. At lower doses (10−7M DEX), GCs promote uncommitted mesenchymal precursors cells to differentiate into osteoblasts [28,29]. However, at high doses (>10−7M DEX), GCs inhibit the proliferation of osteoblast-like cells in culture [30]. GCs decrease bone formation in most in vivo studies but stimulate it in in vitro studies. These opposing effects could be due to low levels of GCs being stimulatory at lower concentrations and inhibitory at higher concentrations [31].
Multipotent mesenchymal precursor cells could develop into osteoblasts, adipocytes or other cell lineages. GCs facilitate the commitment of these precursors cells to differentiate into certain cell types [32]. GCs stimulate the expression of markers of differentiation of both osteoblasts and adipocytes in in vitro studies [33]. In animal and human studies, GCs are associated with bone marrow adiposity within the femoral neck [34,35]. Genetic deletion of 11β-HSD1 and the lowering of GC levels were reported to affect marrow adipose tissue but not bone formation [36].
GCs upregulate DKK1 expressed by the osteoblasts, which in turn suppress anabolic osteoblast behavior [37]. GCs also stimulate osteoblast apoptosis in vitro via the increased endoplasmic reticulum stress through a synergistic pathway with TNFα [38]. This triggers the rapid activation of the kinases Pyk2 and JNK and increases reactive oxygen species in primary cultures [39,40]. The Bcl2 family such as Bim is the proapoptotic factor, and its expression is regulated by GCs [41]. GCs also upregulate Bak expression and downregulate Bcl-xL expression (prosurvival) [42]. DEX can also induce Bcl2-mediated cell death via the induction of p53 [43].
Insulin growth factors (IGFs), transforming growth factors (TGFs), fibroblast growth factors (FGF), and platelet-derived growth factors are also other signalling pathways also targeted by GCs. GCs reduce the anabolic actions of TGFβ and suppress the expression of IGF-I and platelet-derived growth factors, which possess anabolic mitogenic actions in osteoblasts [44,45,46]. Novel cellular targets in osteoblasts that are influenced by GCs in vitro include interleukin (IL)-11 [10], E3 ubiquitin ligases [47], and microRNA-199a [30]. The suppression of IL-11 has been identified as an important mediator of the adverse effects GCs on bone [10]. Therapeutic GCs reduce sex steroid levels [48], which is likely to impact patients with serious inflammatory illnesses greater due to the effects of inflammation on the hypothalamic–pituitary–gonadal axis. This cascade will be harmful to the bone [49]. GC excess leads to central fat accumulation due to its impacts on fat metabolism, which is associated with increased insulin resistance. Obesity and diabetes both have complex impacts on bone metabolism and fracture risk [50,51].
2.2. Action of Glucocorticoid on Oxidative Stress in Osteoblasts
GCs can cause oxidative stress through the production of reactive oxygen species (ROS), downregulation of cytoprotective antioxidant proteins and antioxidant enzyme activities [52]. High ROS levels inhibit osteoblast differentiation and function, causing osteoblast cell death and growth reduction [53,54]. DEX treatment induces oxidative damage by depleting total antioxidant capacity while increasing ROS formation and lipid peroxidation. It also causes a significant reduction in the RUNX2 mRNA expression, which underlies high-dose DEX-induced osteotoxicity [55]. DEX treatment also triggers a significant decline in the mitochondrial membrane potential due to upregulated caspase activity. Treatment with antioxidants can upregulate the expression levels of these osteogenic markers and downregulate caspase expression, thus decreasing the apoptotic effect of DEX. This observation suggests the involvement of oxidative stress in DEX-induced osteoporosis [54].
A study by Zhang et al. [17] found that DEX-treated MC3T3-E1 pre-osteoblast cells express lower nuclear factor (erythroid-derived 2)-like 2 (Nrf-2) and their target proteins with a decline in oxidative stress markers. This negative impact could be reversed with plumbagin, which is an antioxidant. Nrf-2 is a transcriptional activator, which binds to antioxidant responsive element (ARE) and enhances the expression of antioxidant enzymes. This endogenous antioxidant defence is activated in a cellular oxidative stress event. ROS also induces endoplasmic reticulum stress and autophagy-mediated apoptosis [56]. A study by Liu et al. demonstrated that DEX induces apoptosis, endoplasmic reticulum stress, ROS formation and autophagy in pre-osteoblasts [57]. Animal studies demonstrated that high-dose DEX treatment reduced bone generation and destroyed bone trabeculae in rats, leading to microarchitectural degenerative changes mimicking osteoporosis [58,59]. Therefore, inhibiting the production of ROS may provide an avenue of intervention against GC-induced apoptosis of MC3T3-E1 cells [57].
Most in vivo studies showed that GCs usually cause a decrease in bone formation, but some in vitro studies showed largely stimulatory effects of GCs actions. This observation may suggest that a low GC level possesses stimulatory effects, while a high GC level possesses inhibitory effects on bone [10]. GCs exert anti-inflammatory effects on osteoblasts by suppressing cytokines, such as IL-11, via the interaction of the monomeric GR with AP-1 but not nuclear factor kappa B (NF-κB). The inhibition of cytokines by GCs attenuates osteoblast differentiation, which partly accounts for bone loss during GC therapy [60].
2.3. Action of Glucocorticoid on Osteoclasts
GCs create an environment favouring osteoclast formation and bone resorption activities by increasing RANKL and suppressing OPG secretion by osteoblasts [61,62,63,64]. This process may be mediated by miR-17/20a in osteoblasts and miR-182 in osteoclasts [65,66]. GCs also affect osteoclast functions directly. GCs can induce osteoclast-mediated bone resorption without affecting their apoptosis rate, and this process requires the dimeric GC receptor [67]. GCs can improve autophagy in osteoclasts and promote their survival through the PI3K/Akt/mTOR signalling pathway [68]. GCs can affect the geometry of osteoclast resorption activities by forming more trench-like resorption pits, which directly affect bone stiffness, with the lumbar as the most affected bone site [69].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27185862
This entry is offline, you can click here to edit this entry!