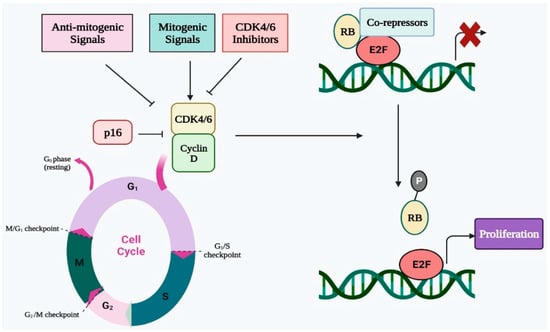
1. Cell Cycle Regulation by CDK4/6 in Normal Cells
Four successive stages make up the cell cycle, which is a highly conserved biological process. These phases are pre-DNA synthesis/G1, S/DNA synthesis, G2/pre-division, and M/cell division. To assure proper progression through the complete cell cycle, many CDKs work with their cyclin partners to govern the transition from one stage to the next. In terms of biochemistry and biology, CDK4 and CDK6 are quite similar to one another, and CDK4/6 can be triggered by D-type cyclins, which are an essential activator of the shift from G1 to S phase [
27,
28]. In the early G1 stage, the number of D-type cyclins begins to rise in response to proliferative stimuli, whereupon these cyclins interact and bind with CDK4/6 and activate them (
Figure 1) [
29]. The cyclin D-CDK4/6 complex, which binds with the transactivation domain of the E2F transcription factor family, thereafter phosphorylates the retinoblastoma (RB) protein [
30,
31]. The E2F transcription factor is released as a result of RB phosphorylation. Additionally, the E2F transcription factor promotes E-type cyclin production before binding to CDK2. The cyclin E-CDK2 complex promotes the G1 to S phase transition while simultaneously accelerating RB phosphorylation and reducing E2F p21 suppression. As a result, CDK4 and CDK6 are essential for the G1 to S phase transition and must be blocked to effectively prevent the G1/S transition [
32,
33]. The cyclin D-CDK4/6 complex becomes hyperactive as a consequence of a rise in cyclin D concentration or CDK4/6 activity, which speeds up the cell cycle and the transition from the G1 to the S phase. Additionally, an accelerated cell cycle leads to unregulated cell multiplication that causes tumor progression [
34]. Thus, CDK4/6 inhibition can result in G1 arrest of the cell cycle, making it a potent and successful approach for cancer therapy. To ensure the effective progression of the cell cycle, cyclin-dependent kinase inhibitors (CKIs) such as INK4 and CIP1/KIP1, regulate CDK activity [
35,
36]. The CDK4/6 function is restricted by INK4 proteins, which either directly attach to CDK4/6 to inhibit its catalytic property or selectively interrupt the interaction of cyclin D with CDK4/6. In contrast to INK4 proteins, the CIP/KIP proteins engage with all CDKs that are essential for the cell cycle and can either suppress or promote CDK activity based upon the cellular environment [
37,
38]. Therefore, the function of CKIs is vital for proper CDK function and healthy cell growth, and cancer may develop as a result of the lack of their function.
Figure 1. Regulatory role of CDK4/6 in cell cycle progression via E2F-Rb pathway.
2. Regulation of Immune Response by CDK4/6
A crucial factor in controlling infections is innate immunity, which acts as the primary defense mechanism for the host. Invasive, fatal infections are linked to deficiencies in innate immunity. Auto-inflammatory conditions can result from improper innate immune system generation. Neutrophils, dendritic cells, macrophages, and innate lymphoid cells are the main cellular elements of innate immunity. The later growth of adaptive immune responses is ultimately regulated by the innate immune system; thus, its appropriate performance is necessary for good health. Cell cycle regulators function primarily as innate immune accelerators during the innate immune response [
39]. Cell cycle molecules such as CDKs and CKIs directly contribute to the proliferation of innate immune system cells and the maintenance of homeostasis in innate immune responses [
40]. Recently, it was discovered that CDKs have a direct, non-cell cycle role in innate immunity. Innate immune cells, such as monocytes, generate type I interferon (IFN) after viral infection to defend the host against viral attacks. The expression of type I IFN (IFN) was suppressed in a monocyte cell line called THP-1 by CDK suppression. It was determined that the activity of CDKs 1, 2, and 4 is necessary for the translation of type I IFN mRNA. IFN-β-mRNA is eliminated from the translational polysome complex in the lack of CDK function directed by a pan-CDK inhibitor such as R547, Dinaciclib, but overall translation is unaffected. This shows that CDK activity is especially necessary for the generation of IFN- β, which in turn triggers immune system activation [
41]. There must be additional follow-up research on these observations. Since the majority of this study’s conclusions were based on pan-CDK inhibitors, genetic tests in which one or more CDKs are selectively knocked out need to be carried out to confirm the findings. Pan-CDK inhibitors may immediately stop the cell cycle, which would have an impact on the translation of the IFN mRNA. The mechanism by which CDK kinase activity maintains the IFN-mRNA in the translating polysome complex is still unknown [
42]. Furthermore, it is unknown how active CDK/cyclin complexes, which are known to be in the nucleus, may regulate cytosolic translation.
Following an initial reaction to a particular pathogen, the adaptive immune system (acquired immune system), develops immunological memory, which results in an improved response to repeated exposures with the infectious agent. The cells that generate the acquired/adaptive immune response are known as lymphocytes. The two main categories of adaptive immunity are antibody and cell-mediated immune responses, which are produced by B and T-lymphocytes, respectively. Genetic studies revealed that particular cell cycle regulators are necessary for the number of adaptive immune cells, as with innate immune cells. Thymocyte counts must be maintained by cyclin D3-CDK6 activity. Mice lacking either CDK6 or cyclin D3 displayed a marked decline in thymocyte counts [
43,
44]. Additionally, cell cycle proteins indirectly regulate lymphocyte functioning during T cell activation. To create effector cytokines in reaction to secondary exposure, T cells must undergo extra cell cycle rounds during the main response [
45,
46]. D cyclins connect the growth and activity of B cells, and subsequently B cell clonal growth entails quick proliferation to produce a germinal core (GC). B cells go through class switching, clonal proliferation, and selection within the GC to produce a high-affinity humoral antibody.
In both mice and human dendritic cells, CDK4 is able to phosphorylate the protein kinase mitogen-activated protein kinase 8 (MAPK8; also known as JNK), which triggers the release of IL-6 and IL-12 in response to transcriptional processes that are dependent on AP-1 [
47]. Furthermore, because neutrophils are terminally differentiated cells, CDK4 and CDK6 have a role in the capability of neutrophils to react against microbial infections by evading neutrophil extracellular traps, a function that is inversely controlled by the broad-spectrum CDK inhibitor p21CIP1 [
48]. These results demonstrate the critical involvement of CDKs in immune system function and development by defining the needs for various CDKs based on cell type and developmental status, as well as a wide range of cell cycle independent functions.
3. Dysregulation of CDK4/6 Activity in Cancer
Mutations and deregulation of many cell cycle regulators, including CDKs, cyclins, CKI, CAK, CDK substrates, and checkpoints, have often been detected in malignant cells [
49]. The fact that CDK4/6 expression levels are substantially elevated in several cancers is well known [
50,
51,
52,
53]. Through both direct and indirect phosphorylation of RB (via inducing CDK2), overexpressed CDK4/6 promote G1/S conversion and promote carcinogenesis. Along with overexpression, CDK4/6 upregulation is a common occurrence in various malignancies (melanoma, leukemia, lymphoma, glioma, sarcoma, etc.) with distinct tissue selectivity for CDK4/6. Compared to its homolog, CDK4 favors mesenchymal tissues (including leukemias and sarcomas), and epithelial tumours (i.e., various malignancies), and some sarcomas tend to express more CDK4 [
54]. The majority of human malignancies contain wild-type RB [
55], and inhibiting hyperactivated CDK4/6 in these cells can stop the cell cycle during G1 stage. Even in RB negative cancers, CDK4/6 inhibitors work by inhibiting cell division or causing apoptosis without involving RB [
56]. Targeting CDK4/6 can also prevent their cell cycle-independent tumor-promoting activities. The modulation of inflammatory cytokine signaling by CDK4 in breast cancer has been revealed using transcriptomic analysis [
57]. CDK6 can activate stem cells, promote angiogenesis, the immune system, and other processes [
58]. In addition, a variety of oncogenes of the signaling pathway promote cell growth by turning on the CDK4/6-RB-E2F pathway. A few examples of these signaling cascades are PI3K/Akt/mTOR, JAK/STAT, RAS/RAF/MEK/ERK, BTK/NF-kB, and Wnt pathways [
59,
60,
61]. Additionally, perturbations in tumour suppressors, such as p53, can trigger the activation of the CDK4/6-RBE2F pathway by relieving the repression of p21CIP1. Thus, CDK4/6 acts as a node in the pathways of carcinogenesis. Despite being optional in normal cells, CDK4/6 have been proven to be essential for the growth of tumour cells in knockout experiments. As a result, it is safe to kill the opposition without endangering allied forces [
62]. Together, these characteristics make CDK4/6 attractive and secure targets for anticancer treatment.
4. CDK4/6 Inhibitors as Immunomodulators in Cancer Cells
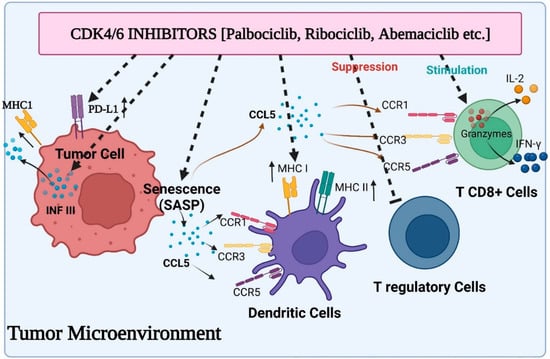
Recent research has shown that CDK4/6 exhibit substantial cell cycle independent activity in tumour immunosurveillance, and CDK4/6 inhibitors may control the immune system response to tumour cells. First, newly available information suggests that CDK4/6 inhibitors may have an immediate immunomodulatory effect on tumour cells (
Figure 2). Abemaciclib treatment in this situation has been associated with the overexpression of multiple antigen processing and antigen presentation machinery elements, including MHC class I molecules (specifically, HLA-A, HLA-B, and HLA-C in human systems, and H2-D1, H2-K1, and B2M in mice systems). OVA-expressing mouse tumour cells become more susceptible to OVA-specific OT-I T cells when exposed to abemaciclib in vitro, and the therapeutic efficacy of abemaciclib in vivo in syngeneic immune-competent animal models is reversed by CD8+ T cell depletion, supporting the idea that MHC class I overexpression plays a mechanistic role in the therapeutic effects of abemaciclib [
63,
64]. In addition, T cells that invade abemaciclib-treated breast cancer have lower amounts of exhaustion biomarkers such as PD1 and cytotoxic T lymphocyte-associated protein 4 (CTLA4). Moreover, the microenvironment of mice colorectal cancers administered with abemaciclib is characterized by elevated expression of transcripts encoding T cell activation signals such as IFN-γ and granzyme B (GZMB), as well as co-stimulatory receptors such as TNF receptor superfamily member 9 (TNFRSF9).
Figure 2. Various aspects of immunomodulation via CDK4/6 inhibitors on the tumor microenvironment: induced activity of antigen presentation and PD-L1 expression on tumor cells, suppressed activity of Treg cells, enhanced cytokine secretion from CD8+ T cells and stimulated function of antigen presentation by dendritic cells.
Following a combination of the PI3K inhibitor BYL719 plus ribociclib or palbociclib, similar outcomes were seen in TNBC models [
65]. Researcher revealed that immune-associated pathways were predominant in cancerous cells after receiving therapy with a medication cocktail by conducting a gene set enrichment experiment. Specifically, the elevation of genes associated with antigen presentation and genes coding for cytokines was noticed, suggesting the immunomodulatory impact of these therapies. In contrast to this, new research has shown that CDK4/6 inhibition may support tumour immune evasion in a number of tumour types through the increase of the immunosuppressive programmed cell death protein 1 ligand (PD-L1) expression on tumour cells. Effector T cell functioning in peripheral tissues is regulated by the PD1/PD-L1 immune checkpoint system [
66], and PD-1 interaction with T cells directly reduces TCR-mediated effector functions, serving as a negative immune response modulator. Because immune-checkpoint inhibitors have recently been developed, and their activity may be correlated with PD-L1 expression on tumour cells, they may be able to counteract the detrimental immune-suppressive effect of CDK4/6 inhibitors. Accordingly, Zhang and colleagues found an inverse relationship between CDK4 activity and PD-L1 expression on cancer cells, and showed that palbociclib or ribociclib treatment increased PD-L1 expression in a variety of in vitro and in vivo models, regardless of RB status, denoting that the RB/E2F axis was not responsible for this in these models. In fact, the researchers concluded that the Cullin 3-based E3 ligase, which coupled with PD-L1 via the adaptor protein specle-type POZ (SPOP), modulated the level of PD-L1 protein. In particular, CDK4/Cyclin D-mediated phosphorylation and alternate interaction with the proteins 14-3-3γ or FZR1 were responsible for controlling SPOP stability. FZR1 destroyed SPOP, which was no longer phosphorylated in the presence of CDK4/6 inhibitors, boosting the expression of PD-L1 [
67]. Palbociclib and an anti-PD-1 antibody treatment significantly prolonged overall survival compared to single medication treatments in mouse models of cancer, which supports the idea that immune-checkpoint antagonists and CDK4/6 inhibitors should be combined to create a more potent anticancer medicine. The enhanced production of PD-L1 by CDK4/6 inhibitors has now been linked to the stimulation of the transcription factor NF-kB in RB-proficient cell lines from various tumour types, as well as an increase in the protein’s durability. It is well known that the NF-kB protein p65 controls the expression of the PD-L1 gene [
68]. The interaction of this protein with hyperphosphorylated RB protein can limit the transcriptional activity of RB protein, which in turn inhibits the expression of the PD-L1 gene. As a result, RB loss and CDK4/6 inhibitors both cause the up-regulation of PD-L1 by encouraging the release of active p65 protein. Recently, it was observed that CDK4/6 activity and PD-L1 expression are correlated in melanoma patients [
69]. In animal models of melanoma, the mixture of CDK4/6 inhibitors and anti-PD-1 antibodies substantially inhibited cancer growth. It is well established now that CDK4/6 inhibition causes SASP in a number of different cell models. Senescent melanoma cells generated by CDK4/6 inhibitors have been shown to generate CCL5 via activation of NF-kB, a member of the released factors [
70]. A chemokine called CCL5 interacts with the CCR5, CCR3, and CCR1 receptors located on a myriad of immune cells, including natural killer cells, immature dendritic, and activated T cells [
71]. Researchers revealed that cytokines secreted by senescent cells encouraged the infiltration of immune cells into the cancer using PDX tumours taken from melanoma patients. They found that the expression of markers linked to T lymphocytes (CD2, CD3, CD8A and B, CXCR3, CCR5) and cytotoxic immune cells was positively correlated with the secretion of CCL5 (granzymes and FAS ligand).
This entry is adapted from the peer-reviewed paper 10.3390/ijms24032236