Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
|
Oncology
Immunotherapy has brought new hope for cancer patients. There is still a need to address major challenges including heterogeneity in response among patients, the reoccurrence of the disease, and iRAEs (immune-related adverse effects). The first critical step towards solving these issues is understanding the epigenomic events that play a significant role in the regulation of specific biomolecules in the context of the immune population present in the tumor immune microenvironment (TIME) during various treatments and responses.
- cancer
- immunotherapy
- immune checkpoint drugs
- Epigenetics
1. Introduction
Immunotherapy (a type of cancer treatment that relies on empowering the body’s own immune cells to fight cancer) has expanded to include: i—immune checkpoint/ligand inhibitors (CTLA-4, PD-1, PD-L1/L2, TIM3, and TIGIT) [1,2,3,4,5,6,7,8,9,10,11,12]; ii—adoptive T-cell transfer therapy (CAR-T, TCR-T, TIL and NK cell) [13,14,15,16,17]; iii—cancer vaccines (T-vec, BCG and Sipuleucel-T) [18,19]; and iv—immunomodulators (thalidomide, lenalidomide and pomalidomide) [20,21,22,23]. Immunotherapy has revolutionized cancer treatment, providing significant clinical benefits to patients with different types of cancers. However, only a small subset of patients benefit from immunotherapy, which highlights limitations of this therapy. Major limitations include the low response rate evidenced by primary/acquired resistance and iRAEs [24,25,26,27]. These limitations can be attributed to epigenetic changes acquired by the TIME that play an imperative role in the development of intra/inter tumor heterogeneity by favoring the evolution of transcriptionally distinct clonal populations of cancer cells, which ultimately aid tumor progression and development [28,29].
Epigenetic aberrations are considered hallmarks of cancer development and progression [30]. In the TIME, cancer cells escape immune-mediated cell death by utilizing epigenetic mechanisms to escape host immune recognition and immunogenicity [31,32]. In the tumor microenvironment (TME), in addition to cancer cells, immune cells also undergo various epigenetic modifications that alter their effector cytokine expression, cancer immunosurveillance, immune-checkpoint molecule expression, and tumor-associated antigen presentation with MHC molecules [33,34]. Additionally, epigenetic modulators such as DNA methyltransferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis) can re-program the TIME to increase the susceptibility of tumor cells to cytotoxic T-cell-mediated killing, leading to enhanced anti-tumor immune responses [35,36]. Moreover, unlike genetic alterations, epigenetic modifiers can be pharmacologically altered to revert the changes acquired during cancer initiation and progression [37,38,39,40].
An improved understanding of epigenetic events related to immunotherapy resistance would be helpful in designing potential combination strategies for immunotherapy. Multiple factors including constitutive PD-L1 expression in cancer cells, a lack of tumor antigens, defective antigen presentation and processing machinery, the exhaustion of infiltrated T cells, and the presence of an immunosuppressive population—such as Tregs, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs)—could contribute to acquired resistance to immunotherapy [41,42,43] for TAMs [44].
2. Epigenetic Modifiers in T Cells
The functional differentiation of T cells, like short-lived effectors, long-term memory T cells, Treg, and other T-cell populations, is majorly influenced by epigenetic modifications. An increasing number of investigations support the crucial role of HATis, HDACis and HMTs in regulating the fate and function of T cells. The inhibition of HDAC1 and HDAC2 promote the differentiation of CD4+ T cells into cytotoxic CD4+ T cells [50,51]. HDAC3 is critical for the maturation of both CD4+ and CD8+T cells and the production of TNF upon TCR/CD28 stimulation [52]. Enrichment in the central memory and stem cell memory phenotypes of T cells is regulated by H3K4me3 modification at specific gene promoters such as TCF7, LEF1, and KLF2. Interestingly, the upregulation of H3K4me3 and the downregulation of H3K27me3 at the Gcnt1 locus were found to enhance the trafficking of memory T cells to tumor sites in an interleukin (IL)-15-dependent manner [53].
Scheer et al. reported that lysine methyltransferase Dot1l-dependent H3K79me2 is crucial for CD4+ T helper (Th) cell differentiation, as the loss of it was found to lead to the increased expression of Th-1-specific genes and the overproduction of IFN-γ at the expense of Th-2 cell development, advocating a central role for Dot1l in Th-2 cell lineage commitment and stability [54]. Another study investigated the role of menin, a major component of the trithorax group (TrxG) using Cd4-cre-driven conditional knockout (KO) mice; a deficiency in menin was shown to lead to the downregulation of Gata3 expression due to reduced levels of H3K9ac and H3K4me3 at the upstream regions of the Gata3 proximal promoter [55]. Interestingly, the suppression of histone H3K27 demethylases KDM6A (UTX) in mature Th-17 cells was found to reduce mitochondrial biogenesis, causing metabolic reprogramming and reducing the expression of key metabolic TFs, such as PPRC1, which ultimately showed anti-inflammatory effects [56]. The results of these studies reinforce the role of epigenomic events in T-cell biology.
2.1. Epigenetic Modifiers in Immune Checkpoint Therapy
A critical balance between immune co-inhibitory and co-stimulatory signals in the TIME is maintained to restrict tumor development and progression (Figure 1) [57,58]. The epigenetically regulated aberrant expression of immune checkpoints (ICs), including PD-1, CTLA-4, TIM-3 (T-cell immunoglobulin and mucin-domain containing-3), LAG-3 (lymphocyte-activation gene 3), TIGIT (T-cell immunoreceptor with Ig and ITIM domains), VISTA (V-domain Ig suppressor of T-cell activation), CD276 (B7-H3), B7-H4 (VTCN1/B7x/B7S1/B7 homolog 40), IDO-1 (indoleamine 2,3-dioxygenase 1), CD161, CD38, CD93, and CD47 may result in the induction of an immune-suppressive environment, which helps tumor cells to evade immune destruction [12,59,60]. Targeting altered epigenetic modifications can significantly contribute to the reversal of the transcriptomic regulation of ICs and their ligands, which could help to re-establish potent host immunosurveillance mechanisms [61].

Figure 1. Interaction of co-stimulatory/inhibitory molecules between T cells and APCs/tumor cells provides an overview of the immune checkpoint/stimulatory molecules involved in the anti-tumor immune response.
DNMTis and HDACis have been shown to cause the upregulation of immune-signaling components and antigen presentation through the expression of ERVs (endogenous retroviral sequences), thereby improving tumor cell recognition [62]. Decitabine (DNMTi) upregulates the cancer testis antigen member MAGE-1 via hypomethylation, thus increasing chances of its presentation through MHC molecules to effector immune cells [63]. Panobinostat (pan HDACi) has been found to significantly increase CD38 expression in multiple myeloma, so it has been utilized in the development of an effective combinatorial treatment with daratumumab [64]. Panobinostat also affects the PD-L1/PD1 axis via the upregulation of PD-L1 in melanoma cells, which can be then targeted with anti PD-L1 antibodies [65].
H3K9 lysine methyltransferase, SETDB1, has a critical role in the carcinogenesis of multiple tissue types through the transcriptional silencing of multiple genes at specific loci. The amplification and increased expression of SETDB1 in advanced clear renal cell carcinoma has been found to be associated with a poor response to anti-PD1 therapy [66]. Further exploring this information, the Bernstein lab identified that the reversal of the epigenetic silencing of SETDB1 activates tumor immunogenicity through the hypomethylation of H3K9 in the transposable elements that reside in the MHC peptidome [67,68]. The results of these studies indicate the high potential of SETDB1 inhibitors such as mithramycin in combination with immune checkpoint blockade therapy (ICT) [69].
The H3K27Ac reader bromodomain and extra-terminal motif (BET) protein is overexpressed in various cancers and involved in the regulation of the (PD-1/PD-L1) immune checkpoint axis [36]. Accordingly, targeting BET with JQ1 inhibitors in combination with anti-PD1 therapy has been proven to be effective in ovarian and triple-negative breast cancer [36,70]. In another study, Adeegbe et al. showed that JQ1 treatment significantly lowered PD-L1 expression in tumor cells, which led to an increased tumor infiltration of cytotoxic T cells in a non-small cell lung cancer NSCLC xenograft, and a combination treatment of JQ1 with anti-PD-1 reduced tumor burden and resulted in an improved survival rate [71].
The promoter hypomethylation of LAG3 has been a major epigenetic regulator of mRNA expression in clear cell renal cell carcinoma (KIRC), which has a proven association with increased immune cell infiltration and an interferon-γ signature [72]. Beyond cancer, patients’ aberrant histone methylation in chronic osteomyelitis is related to the higher expression of LAG3 in the T cells of peripheral blood [73]. Another negative stimulatory molecule, Tim-3, has been shown to be epigenetically regulated, so its increased expression inhibits the expansion of Th1 and Th17 responses via its binding to galectin-9, ultimately leading to immune exhaustion in the tumor microenvironment [74,75,76]. EZH2-H3K27me3/DNMT3A-DNA methylation regulates the expression of Tim-3 and galectin-9 in HPV18-associated cervical cancer [77]. Tim-3 and galectin-9 are overexpressed in cervical cancer cases, which is mediated through the hypomethylation of HAVCR2 and LGALS9 because of the lesser expression and recruitment of DNMT3A to their promoter regions. SUV39H1, a H3K9me3-specific histone methyltransferase, contributes to Tim-3 and galectin-9 regulation by upregulating the H3K9me3 level at the DNMT3A promoter region, hence downregulating its expression. Therefore, SUV39H1 can be utilized as a potential therapeutic target that can downregulate the immune checkpoint inhibitors Tim-3 and galectin-9 [78].
Another immune checkpoint, TIGIT, was found to be upregulated during T-cell and NK-cell exhaustion [9]. Moreover, TIGIT was reported to be regulated by promoter demethylation in melanoma, thus making it sensitive to anti PD-1 therapy [79].
2.2. Epigenetic Modifiers in Antigen Processing and Presentation
In a proper functioning immune system, T cells recognize tumor antigens based on the binding of a T-cell receptor (TCR) and a matching antigen packaged into major histocompatibility complex (MHC) proteins on APCs. Tumor cells escape immune recognition through multiple mechanisms such as alterations in antigen presentation and processing machinery (APM) or alterations in MHC class I molecules, which further impair their identification by CTLs (Figure 2).
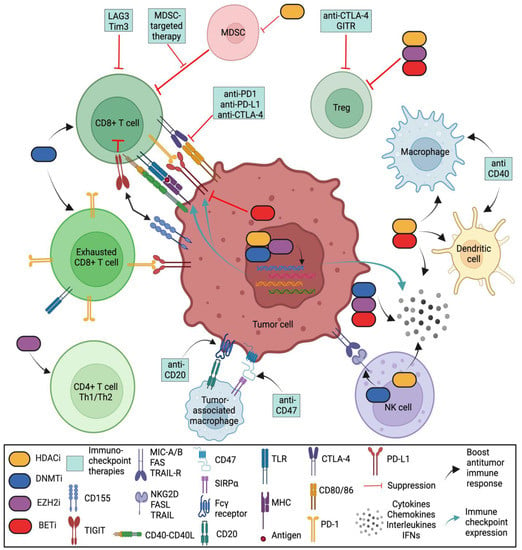
Figure 2. Role of various epigenetic modifiers in the tumor immune microenvironment. DNA methyltransferase inhibitors (DNMTis), histone deacetylase inhibitors (HDACis), an inhibitor of histone methylation on histone H3 at lysine 27 (EZH2i), and inhibitor of bromodomain and extra-terminal motif (BETi) shape the tumor-immune microenvironment by (i) increasing the number of CD8 and CD4 T cells; (ii) activating antigen processing and presentation machinery; (iii) decreasing the abundance of MDSCs and tumor-associated macrophages (TAMs); (iv) downregulating the immune checkpoint inhibitors Tim-3, Lag-3 and TIGIT; (v) upregulating immune checkpoint PD-L1 (by DNMTis, HDACis and EZH2i) and downregulating PD-L1 (by BETi); (vi) enhancing NK-mediated lysis (by HDACis) or decreasing NK cytotoxicity (by EZH2i); and (vii) upregulating inflammatory genes and pathways that control the secretion of interferons (IFNs), cytokines, and chemokines from tumor cells. (Regulatory T cells (Tregs) are a specialized subpopulation of T cells that act to suppress the immune response, thereby maintaining homeostasis and self-tolerance. It has been shown that Tregs are able to inhibit T-cell proliferation and cytokine production, as well as play a critical role in preventing autoimmunity. Tumor-associated macrophages (TAMs) are the key cells that create an immunosuppressive tumor microenvironment (TME) by producing cytokines, chemokines, and growth factors and by triggering the inhibitory immune checkpoint proteins release in T cells. Natural killer (NK) cells are effector lymphocytes of the innate immune system that control several types of tumors and microbial infections by limiting their spread and subsequent tissue damage. Cancer-associated fibroblasts (CAFs) are one of the most abundant and critical components of the tumor mesenchyme; they not only provide physical support for tumor cells but also play a key role in promoting and retarding tumorigenesis in a context-dependent manner. Recent studies have revealed their roles in immune evasion and poor responses to cancer immunotherapy).
An efficient cancer immunotherapy depends on the recognition of antigens loaded onto the MHC molecules of antigen-presenting cells by T cells in the TIME. The epigenomic regulatory factors that can influence the T-cell recognition of tumor antigens include: (1) the aberrant expression of genes involved in the processing or presentation of tumor antigens and (2) the aberrant expression of antigens. There is a subclass of cancer testis antigens (CTAs), including MAGE (melanoma-associated antigen), PRAME (preferentially expressed antigen of melanoma) and NY-ESO-1 (New York esophageal squamous cell carcinoma-1), which are controlled by DNA methylation and remain silenced in mature somatic cells but are demethylated and overexpressed in various cancers [80,81]. Guadecitabine (SGI-110) and decitabine, which are hypomethylating drugs, have been shown to upregulate/overexpress CTAs such as NY-ESO-1 in epithelial ovarian cancer cells and xenografts when used in combination with NY-ESO-1 vaccine and doxorubicin chemotherapy; T-cell responses to NY-ESO-1 have been observed in most studied patients [82,83].
Studies have evidenced that DNMTis and/or HDACis could alter the expression of MHC class I molecules in cancer cells such as neuroblastoma, cervical, and prostate cancer [84]. Furthermore, the expression of different components of the APM pathway such as TAP-1, TAP-2, LMP2, LMP7 and tapasin can be manipulated by both DNMTis and HDACis in different tumor types [85,86,87]. DNMTis and HDACis can regulate the expression of the costimulatory molecules ICAM-1, CD40, CD80, and CD86 [86,88,89].
Histone methyltransferase SETDB1, which maintains heterochromatin (H3K9me3), plays crucial roles in the carcinogenesis of multiple tissue types through the transcriptional silencing of multiple genes [90]. Accordingly, the inhibition of SETDB1 was found to enhance specific cytotoxic T-cell responses against tumors via the activation of immunostimulatory genes, the encoding of retroviral antigens, and the generation of neoantigen MHC-I peptides, thus suggesting that SETDB1 has high potential to synergize with ICT [91]. The HDAC-1/3 inhibitor entinostat, upon combination with a PD-1 axis blockade, was found to lead to the complete remission of tumors, the expansion of neoantigen-specific T cells, and the induction of long-term immunologic memory in immune-competent bladder cancer mouse models [92].
2.3. Epigenetic Modifiers in Tumor-Infiltrating Immunosuppressive Cells
Tumor-infiltrating immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and cancer-associated fibroblasts (CAFs) inhibit T cells’ effector functionality and anti-tumor responses, which lead to the immune escape of tumors. The presence of an immunosuppressive cell population in the TIME could be a major contributory factor in ineffective ICTs [93]. HDACis have antitumor effects in that they reduce the number of MDSCs through various mechanisms of action such as CG-745, a class I–IIb HDACi that induces the infiltration of lymphocytes by increased antigen presentation and that decreases the amount of MDSCs by decreasing the polarization of M2 macrophages in tumors [35]. Valproic acid (VPA), a class-I HDACi, attenuates the immunosuppressive function of MDSCs by downregulating the expression of retinoblastoma 1 (Rb1), toll-like receptor 4 (TLR4), programmed cell death 1 ligand (PD-L1), and interleukin-4 receptor-alpha (IL-4Ra)/arginase [94]. Moreover, the combinatorial treatment of VPA and anti-PD-1 antibodies was found to repress the growth of B16F10 and EL4 tumor models by impairing tumor-infiltrating M2-MDSC accumulation in the tumor microenvironment compared with their individual therapies [95]. Thus, treatment with epigenetic modifiers inhibits MDSC accumulation, thereby augmenting immune checkpoint inhibitors for successful cancer treatment. Vorinostat (suberoylanilide hydroxamic acid, SAHA), a class I–II–IV HDACi, was shown to have anti-tumor potential for a 4T1 mammary mice model in which it decreased MDSC accumulation in the spleen, blood, and tumor while promoting the activation and function of CD8+ T cells [96].
Tregs play significant roles in inducing variety of immune responses, as determined by the expression of Foxp3, a transcription factor in natural Tregs (nTregs) in the thymus [97,98]. Extrinsic molecular signals including IL-2 and TCR, along with a network of transcription factors, are critical for regulating the expression of Foxp3 through epigenomic modulation, which ultimately determines a Treg’s phenotypic plasticity [99,100]. Epigenetic modifiers such as DNMT1 and DNMT3b are differentially bound to Foxp3 promoter and enhancer sites in nTregs compared with extrinsically induced Tregs. Importantly, DNMTis demethylate and activate the Foxp3 promoter and enhancer elements to induce Foxp3 expression and subsequently enable the induction of Foxp3-dependent, Treg-restricted sets of genes [101]. Demethylation in synergy with TGF-β transforms naive T cells into Tregs with high Foxp3 expression and potent, stable suppressive function [102].
Foxp3 expression in Treg cells was found to be significantly upregulated upon treatment with trichostatin-A (TSA), a HDACi [103]. Moreover, the CTLA4, PD-1, GITR and IL-10 genes are reportedly upregulated by TSA [104]. Ohkura et al. reported that Treg maturation, Treg-specific gene expression, and Treg-specific immunosuppressive activity involve epigenetic regulation through genome-wide CpG DNA hypomethylation pattern [105]. In other study, Wang et al. showed that the inhibition of EZH2, a histone-lysine N-methyltransferase enzyme, resulted in Treg-mediated pro-inflammatory activities in the TME, supporting the idea of the generation of an effector T-cell-mediated anti-tumor immune response [106].
This entry is adapted from the peer-reviewed paper 10.3390/cells12030365
This entry is offline, you can click here to edit this entry!