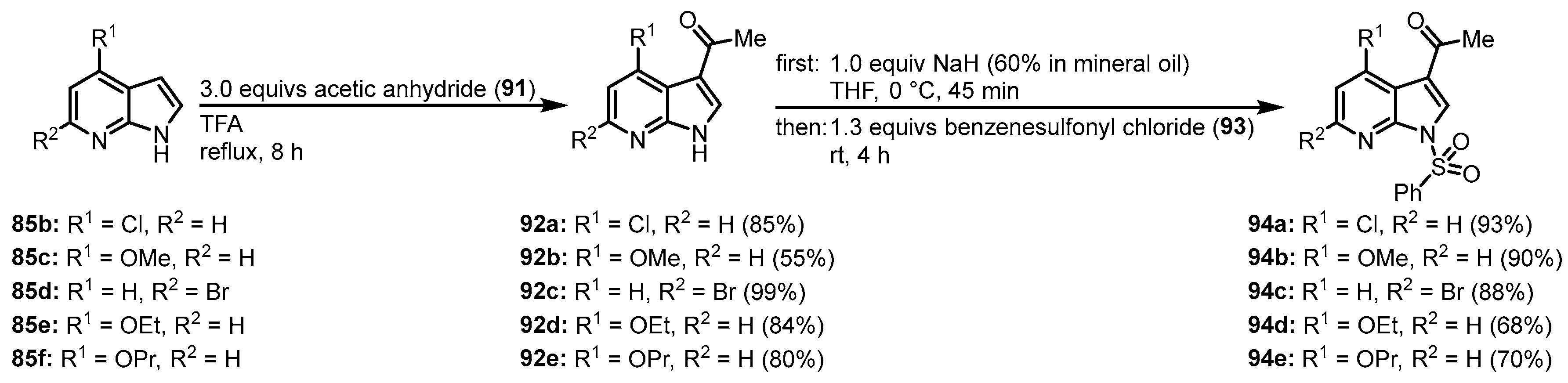1.1. First Total Synthesis by Morris and Anderson
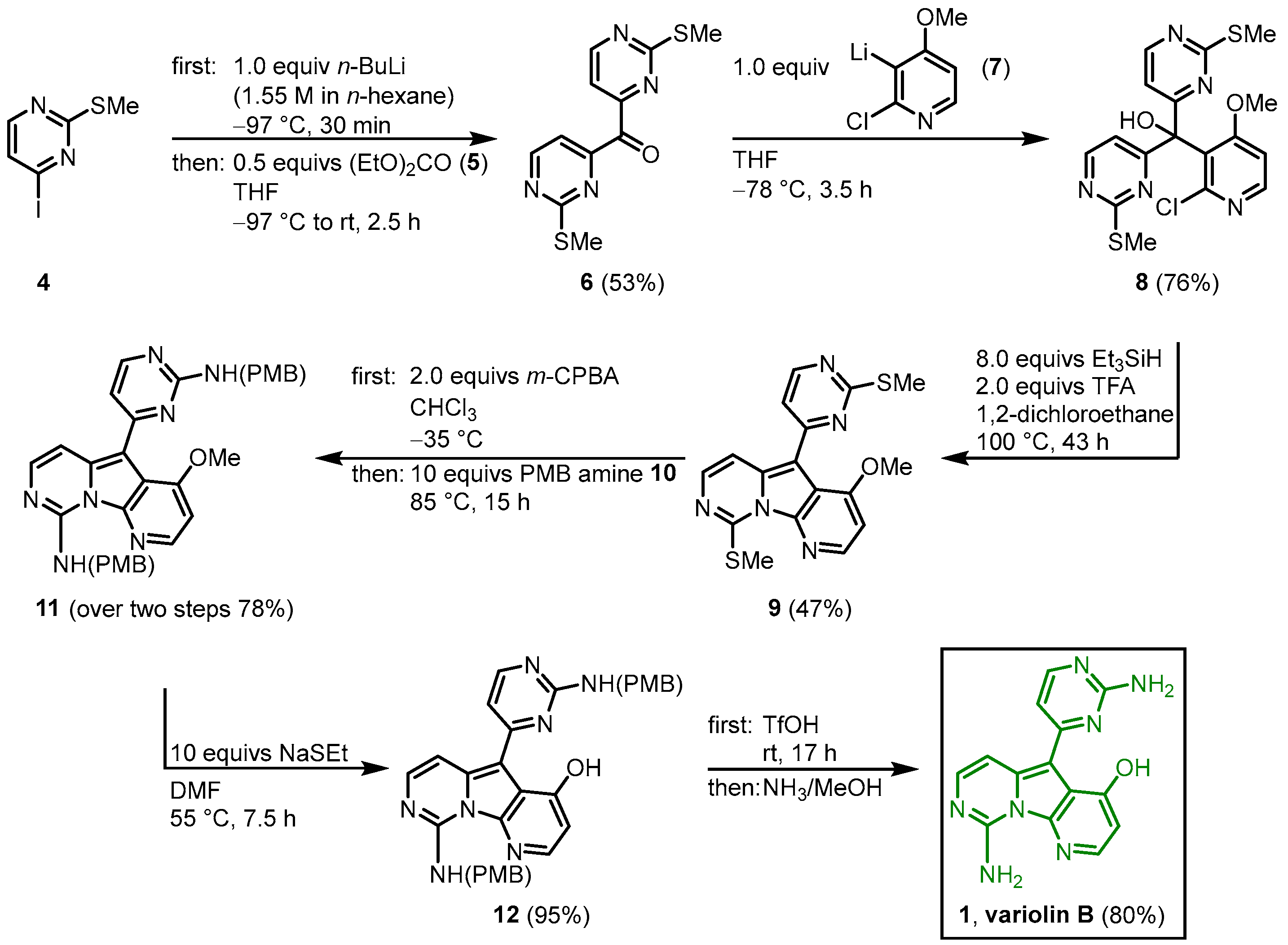
The first total synthesis of variolin B (
1) was achieved by Morris and Anderson in 2001
[1]. Later in 2005, they published the full details of their synthetic strategy together with the synthesis of the synthetic analog desoxyvariolin B
[2]. They recognized the C2-symmetry of intermediate
8, which is cyclized to the pyridopyrrolopyrimidine in the following key step. After halogen lithium exchange in the methylthiopyrimidine
4, the reaction with diethyl carbonate (
5) gave the symmetric ketone
6. The reaction with the lithiated pyridine
7, followed by the key step tandem deoxygenation and cyclization in the presence of triethylsilane and TFA led to the variolin core structure
9. The introduction of the amino groups was achieved by oxidizing the dimethylthiol
9 with
m-chloroperbenzoic acid (mCPBA) to the corresponding disulfoxide, which was reacted with
p-methoybenzylamine (PMB amine) (
10) to give the bisprotected amine
11. Demethylation of
11 and removal of the PMB protecting groups gave the trifluoroacetate salt of the title compound, which was neutralized with concentrated ammonia to give variolin B (
1) in an eight-step synthesis and an overall yield of 11% (
Figure 1)
[1].
Figure 1. First total synthesis of variolin B (
1) by Morris and Anderson
[1].
1.2. Synthesis by Molina and Fresneda
The next synthetic approach was conducted by Molina and Fresneda, who published their syntheses of
1 in 2002
[3] and a modified synthetic route together with the synthesis of an analog in 2003
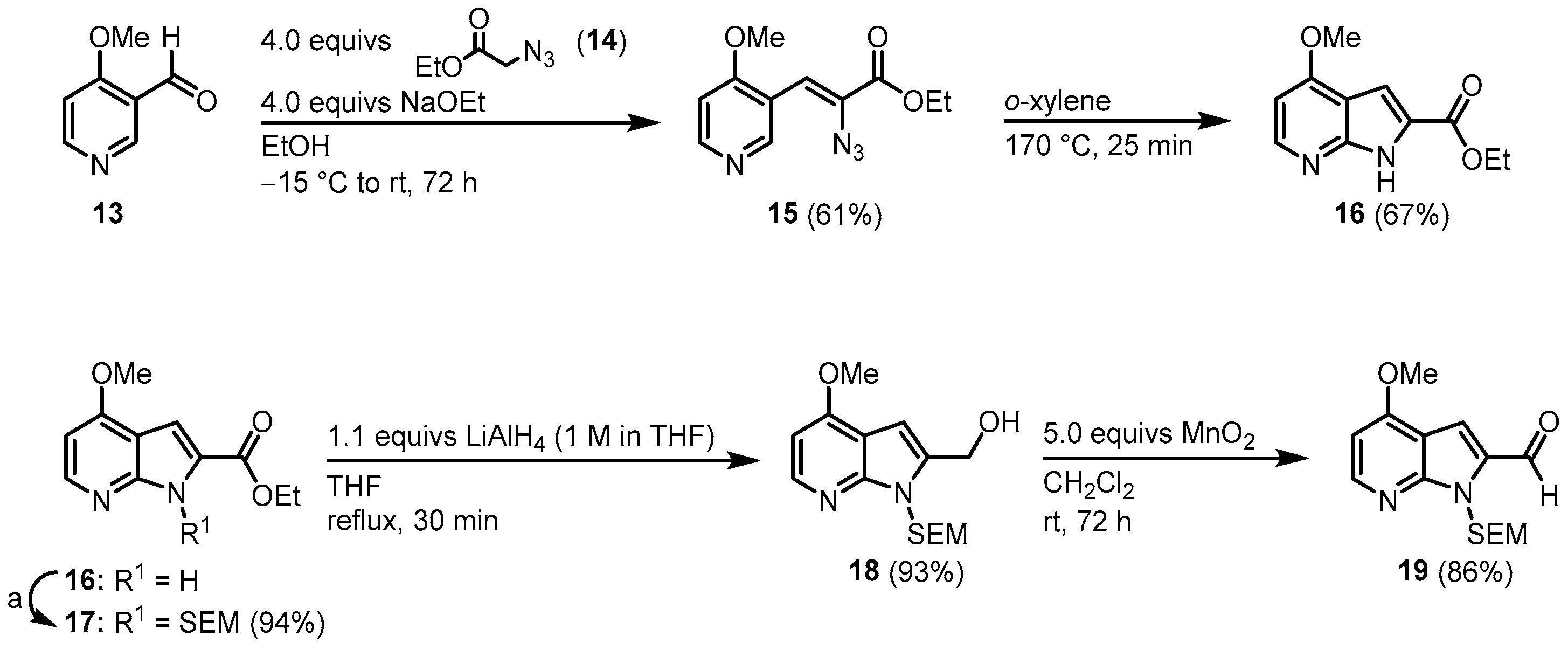
[4]. This approach starts with the synthesis of the 7-azaindole
16. Aldehyde
13 was condensed with azidoacetate
14 and the resulting vinyl azide
15 cyclized to azaindole
16 via a nitrene insertion. After
N-protection with 2-(trimethylsilyl)ethoxymethyl (SEM), the chloride key intermediate
19 was synthesized in a two-step procedure (
Figure 2).
Figure 2. Synthesis of key intermediate azaindole
19. Reaction conditions for a: first: 1.4 equivs NaH, DMF, rt, 45 min. Then: 1.4 equivs SEM-Cl, rt, 12 h
[3].
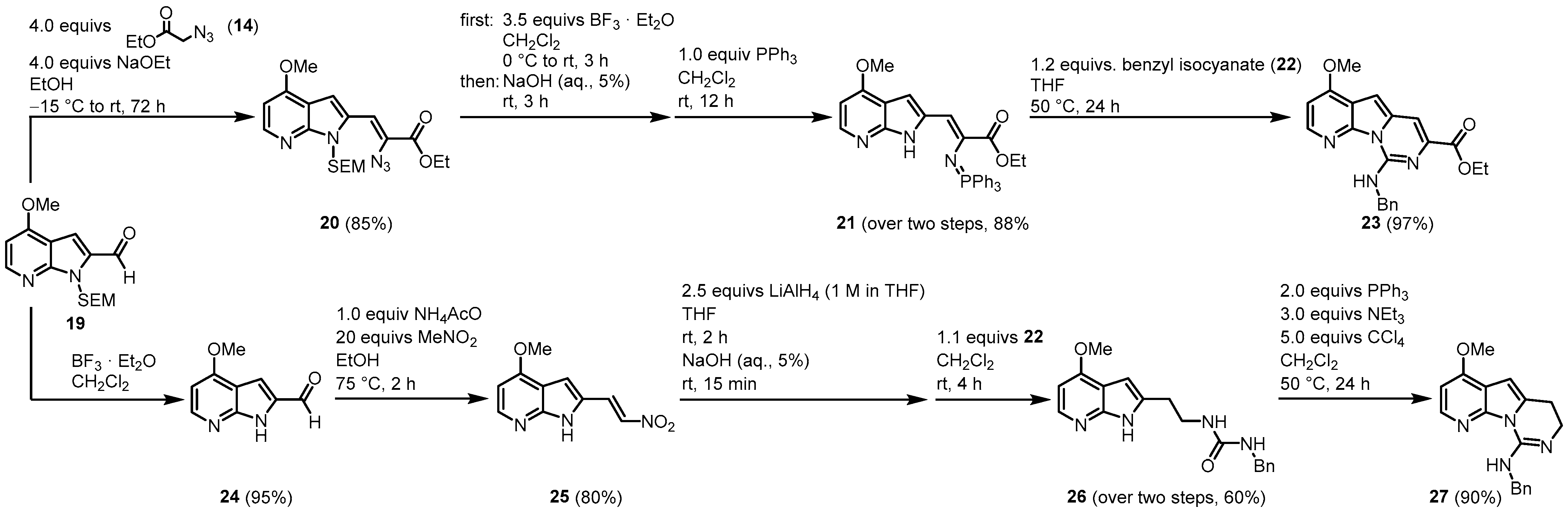
Next, two different approaches are reported (Figure 3). Aldehyde 19 was similarly condensed as aldehyde 13 to give vinyl azide 20. After N-SEM-deprotection, a Staudinger reaction with triphenylphosphane led to iminophosphorane 21 in a one-pot reaction. Reaction with benzyl isocyanate (22) in the key aza-Wittig reaction gave a non-isolable carbodiimide that subsequently cyclized to the desired pyridopyrrolopyrimidine moiety 23.
Figure 3. Two approaches to the synthesis of the tricyclic pyridopyrrolopyrimidine structures,
23 and
27 [3].
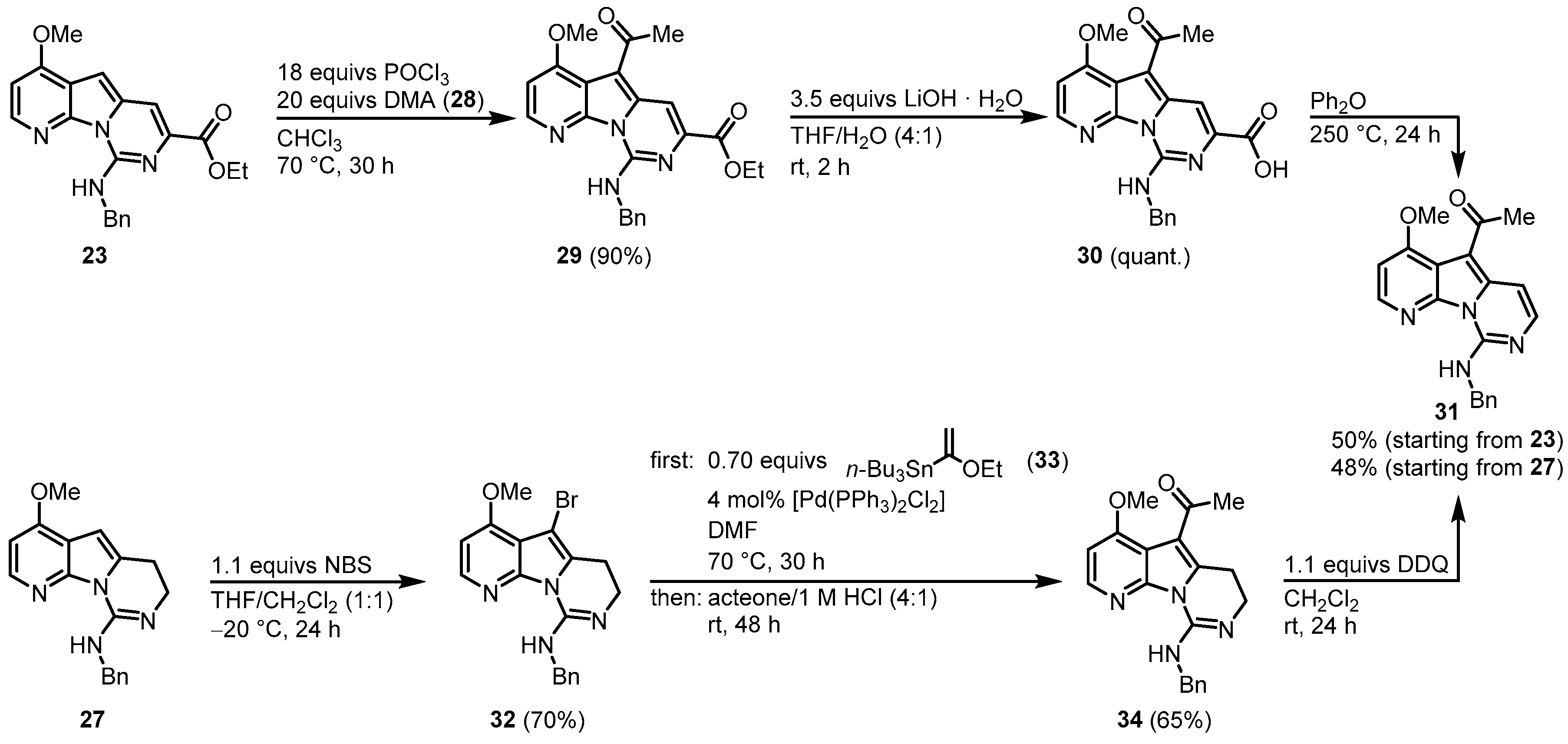
Molina and Fresneda developed a second approach to obtain the tricyclic variolin core without the ester group at C-7. After the N-SEM-deprotection of 19, a nitroaldol condensation with nitromethane led to the formation of 25. Treatment with lithium aluminum hydride gave the corresponding 2-(2-aminoethyl)-7-azaindole, which was sequentially converted to the urea derivative 26 with benzyl isocyanide (22) without isolation. The 26 was dehydrated to the carbodiimide, which subsequently cyclized to the dihydropyrimidine 27 using the Appel reagent (CCl4/PPh3/NEt3). Applying both synthetic approaches, an oxygen substituent is placed at C-4 and a nitrogen substituent at C-9. The next step was to introduce the 2-aminopyrimidine ring at C-5, consequently leading both approaches to the acylated intermediate 31. The reaction of 23 with phosphorus oxychloride and N,N-dimethylacetamide (DMA) (28) allowed the direct introduction of an acetyl group at C-5. Ester hydrolysis led to the carbonic acid 30, and the thermal treatment forced the formation of intermediate 31 by decarboxylation. The route starting from 27 began with the introduction of a bromine substituent at C-5 and the reaction of bromine 32 with n-tributyltin(1-ethoxyvinyl)stannane (33) in the presence of dichlorobis (triphenylphosphine)-palladium(II) introduced to the acetyl group at C-5. Oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave the intermediate 31 (Figure 4).
Figure 4. Introduction of an acetyl group at C-5
[3].
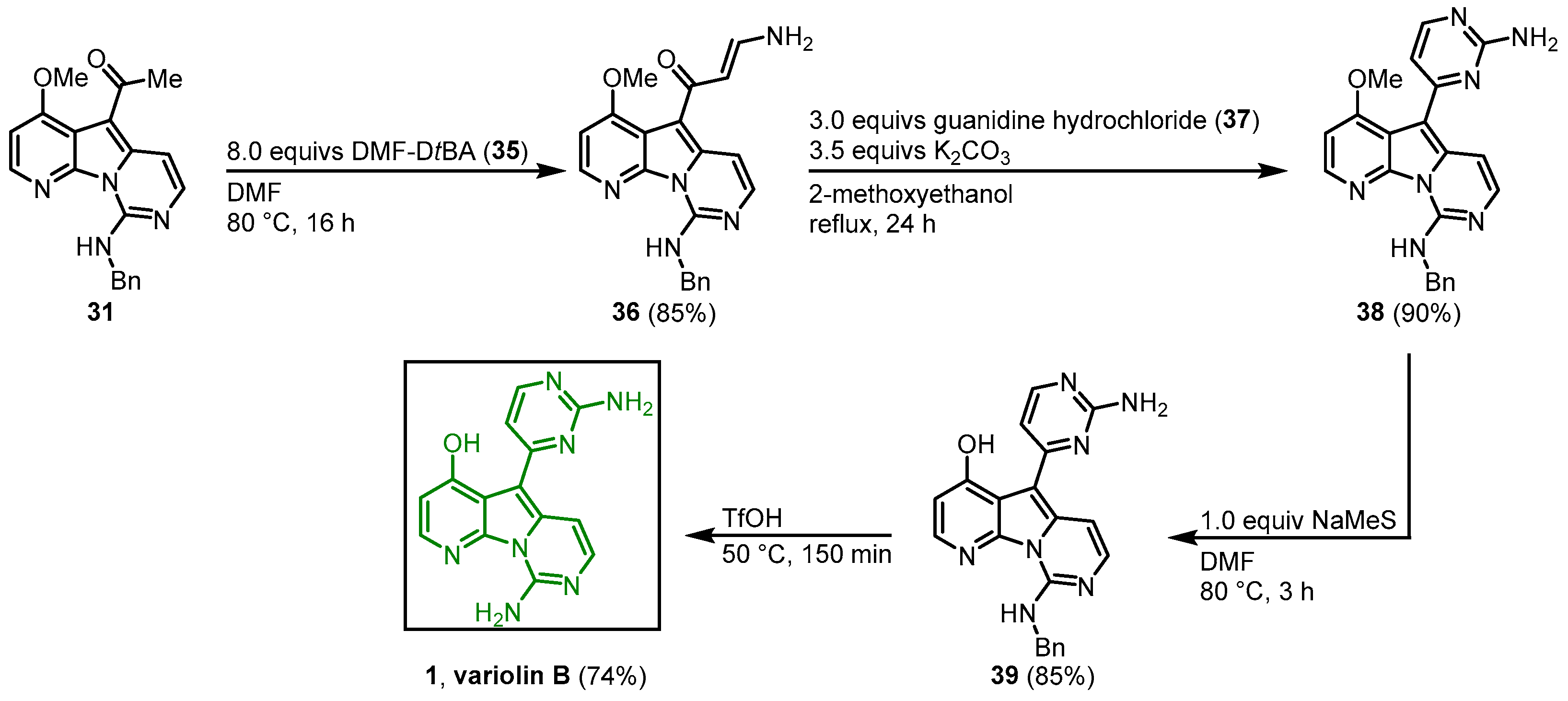
The 2-aminopyrimidine substituent was synthesized using a protocol developed by Bredereck (
Figure 5)
[5]. Enaminone
36 was synthesized from
31 with
N,
N’-dimethylformamide di-
tert-butylacetal (
35) in DMF. Condensation with guanidine hydrochloride (
37) led to ring closure and formed the desired 2-aminopyrimidine
38.
Figure 5. Synthesis of the 2-aminopyrimidine ring to give access to variolin B (
1)
[3].
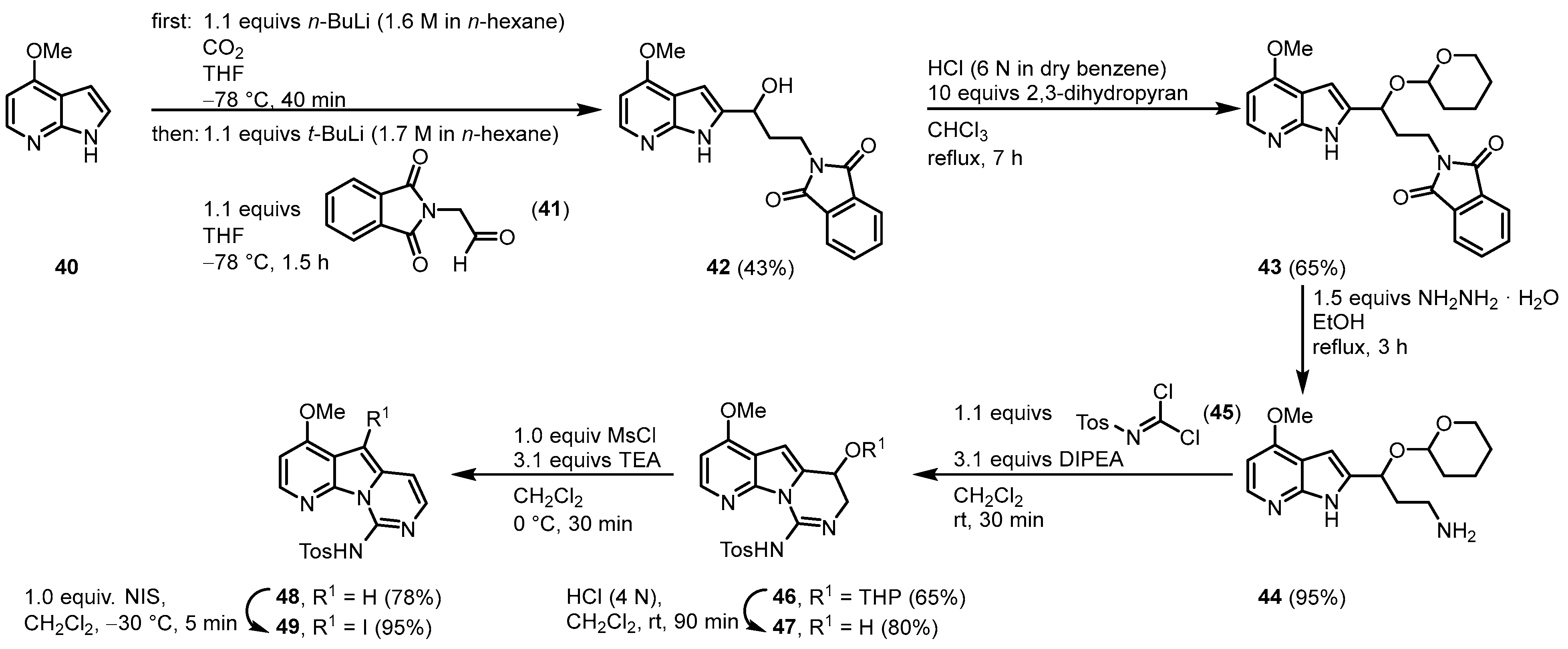
1.3. Variolin B Approach by Alvarez
In 2003, Alvarez published the synthesis of variolin B (
1) and the synthetic analog desoxyvariolin B
[6][7][8]. Starting from 4-methoxy-7-azaindole (
40), a lithium carboxylate was used as an
N-protecting group as well as an
ortho-directing substituent to form a 2-lithio-7-azaindole with a protocol by Katritzky
[9]. Reaction with 2-(1,3-dioxoisindolin-2-yl)acetaldehyde (
41) gave the alcohol
42 that was protected with dihydropyran.
N-deprotection of
43 by hydrazinolysis gave the aminoacetal
44. Ring closure was achieved by the reaction with
N-tosylcarbonimidic dichloride (
45) and diisopropylethylamine (DIPEA) giving
46 in a diasteriometric mixture in a ratio of 1:1. Removal of the
O-tetrahydropyran (THP) protecting group and elimination of the resulting hydroxy group by the formation of its mesylate and treatment with triethylamine afforded the pyridopyrrolopyrimidine scaffold (
48). Regioselective iodination with
N-iodosuccinimide (NIS) gave the key intermediate
49 (
Figure 6).
Figure 6. Synthesis of the key intermediate iodide
49 [6].
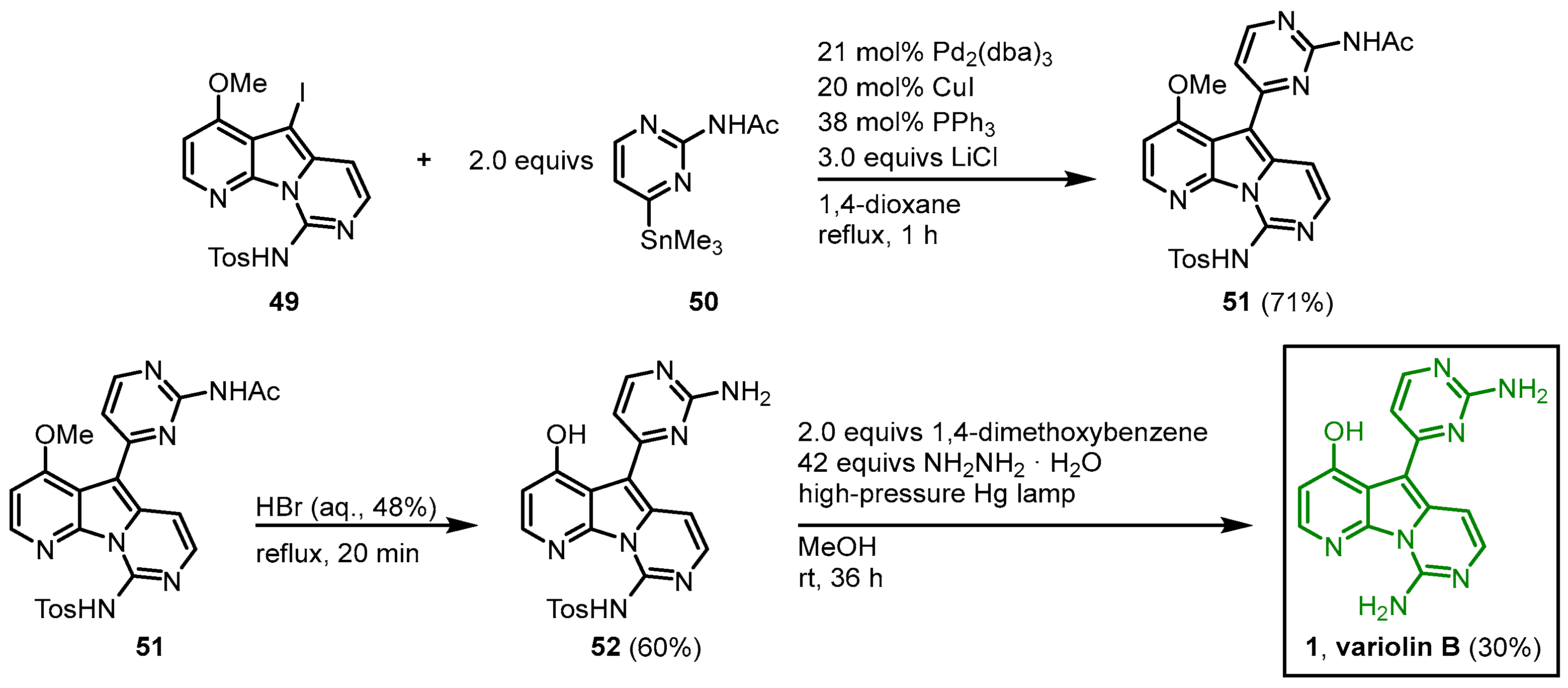
A Stille reaction of 49 and 2-acetylamino-4-trimethylstannylpyrimidine (50) in the presence of tris(dibenzylideneacetone)dipalladium(0) afforded 51. The O-demethylation and N-acetyl-deprotection were achieved by the treatment of 51 with hydrobromic acid, and after reductive photolysis with hydrazine as a reducing agent and 1,4-dimethoxybenzene as an electron source, the tosyl group was cleaved to give variolin B (1) in a 10-step synthesis with an overall yield of 1% (Figure 7).
Figure 7. Synthesis of variolin B (
1) via Stille coupling as a key reaction step
[6].
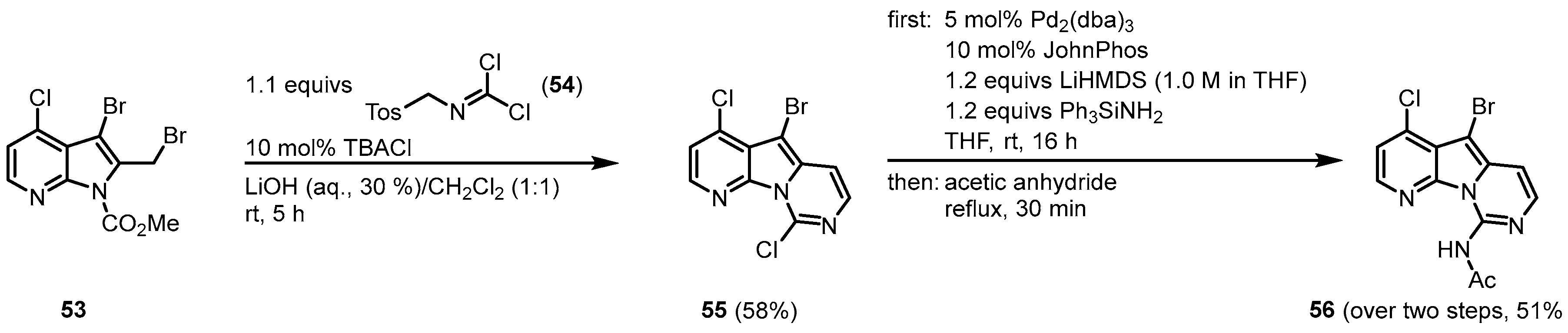
1.4. Synthesis of Variolin B by Burgos and Vaquero
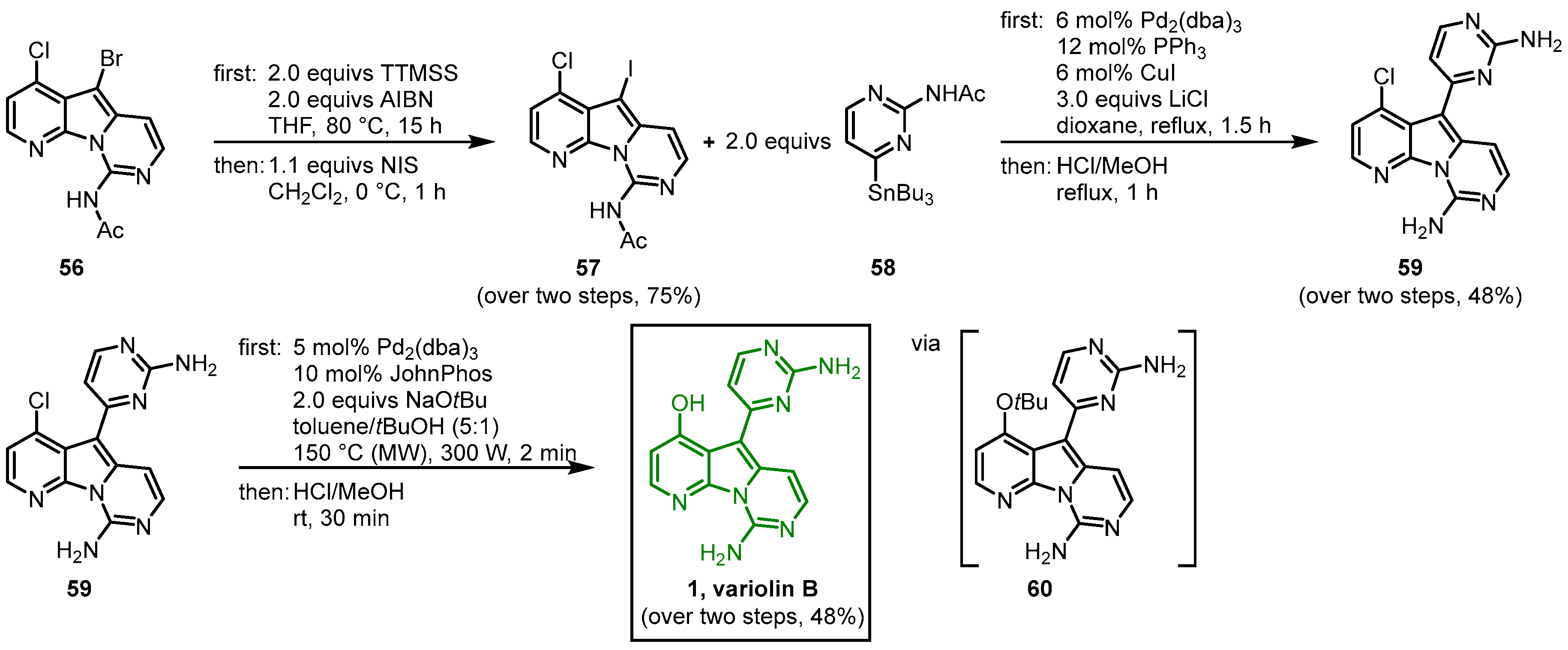
The 2008 approach by Burgos and Vaquero to synthesize variolin B (
1) followed the strategy to design the highly functionalized trihalo-substituted pyridopyrrolopyrimidine core
55 and introduce the substituents via palladium-mediated cross-coupling reactions
[10][11]. The functionalized 7-azaindole
53 was synthesized from 7-azaindole in six single steps
[12]. The
53 was reacted with
N-tosylmethyl dichloroformimide (
54) under phase-transfer conditions in the two-phase system LiOH (aq., 30%)/CH
2Cl
2 (1:1) with tetrabutylammonium chloride to give the trihalo-substituted compound
55. The C-9 amino substituent was introduced by a palladium-mediated C-N bond formation, using lithium bis(trimethylsilyl)amide (LiHMDS) and triphenylsilylamine as an ammonia source. The reaction required the use of the ligand [1,1′-biphenyl]-2-yldi-
tert-butylphosphane (JohnPhos). After
N-acetyl-protection,
56 was obtained (
Figure 8).
Figure 8. Synthesis of the trihalo core and introduction of the C-9 amino substituent
[10].
Next, in a debromination-iodination process, tris(trimethylsilyl)silane (TTMSS) and azobisisobutyronitrile (AIBN) and subsequently NIS were used to exchange the bromo compound
56 to the more reactive iodo derivative
57. In a palladium-catalyzed cross-coupling reaction with the pyrimidyl stannyl reagent
58, the C-C bond at C-5 was formed and the deprotection of both amino groups led to
59. Then, in a palladium-promoted C-O coupling microwave (MW) reaction with sodium
tert-butoxide, the
tert-butyl group was introduced at C-4 to give the
tert-butyl ether
60, and in a final step, the
tert-butyl moiety was cleaved to give variolin B (
1) (
Figure 9). Starting from
53, variolin B was synthesized in seven steps with an overall yield of 5%
[10][11].
Figure 9. Palladium-mediated synthesis of variolin B (
1)
[10].
2. Syntheses of Meridianins
2.1. First Total Synthesis of Meridianins D and G by Jiang and Yang
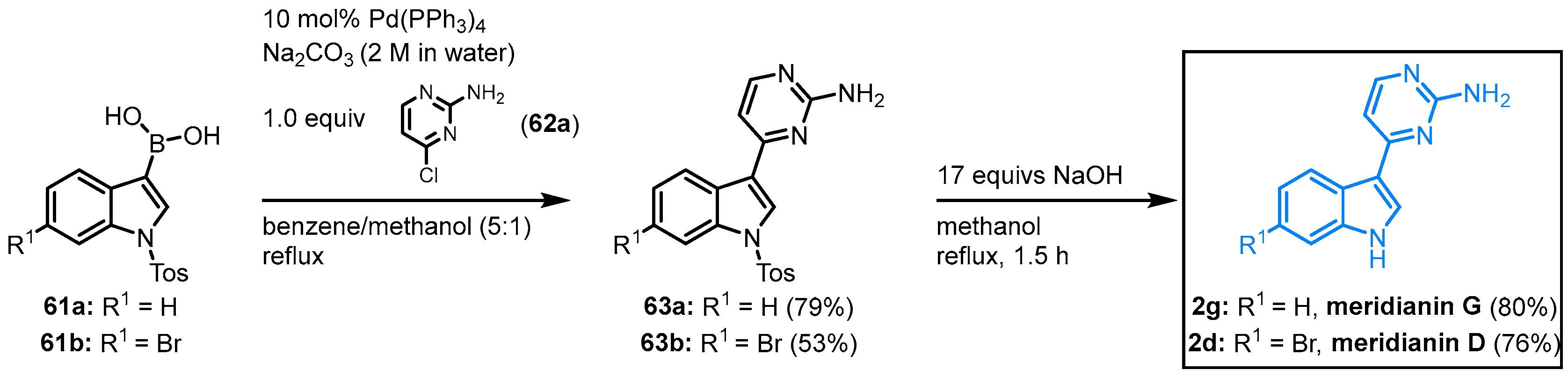
In the early 2000s, Jiang and Yang published a straightforward synthesis of meridianins D and G. Starting from the corresponding indolyl boronic acid,
61 with 4-chloropyrimidine
62a is the key reaction in this meridianin synthesis to furnish protected meridians
63 (
Figure 10). After
N-tosyl-deprotection of compounds
63 with sodium hydroxide, meridianin G (
2g) is obtained in an overall yield of 63%, and meridianin D (
2d) in an overall yield of 40% in this two-step synthesis
[13].
Figure 10. First synthesis of meridianins D and G
[13].
2.2. Synthesis of Meridianins by Fresneda and Molina
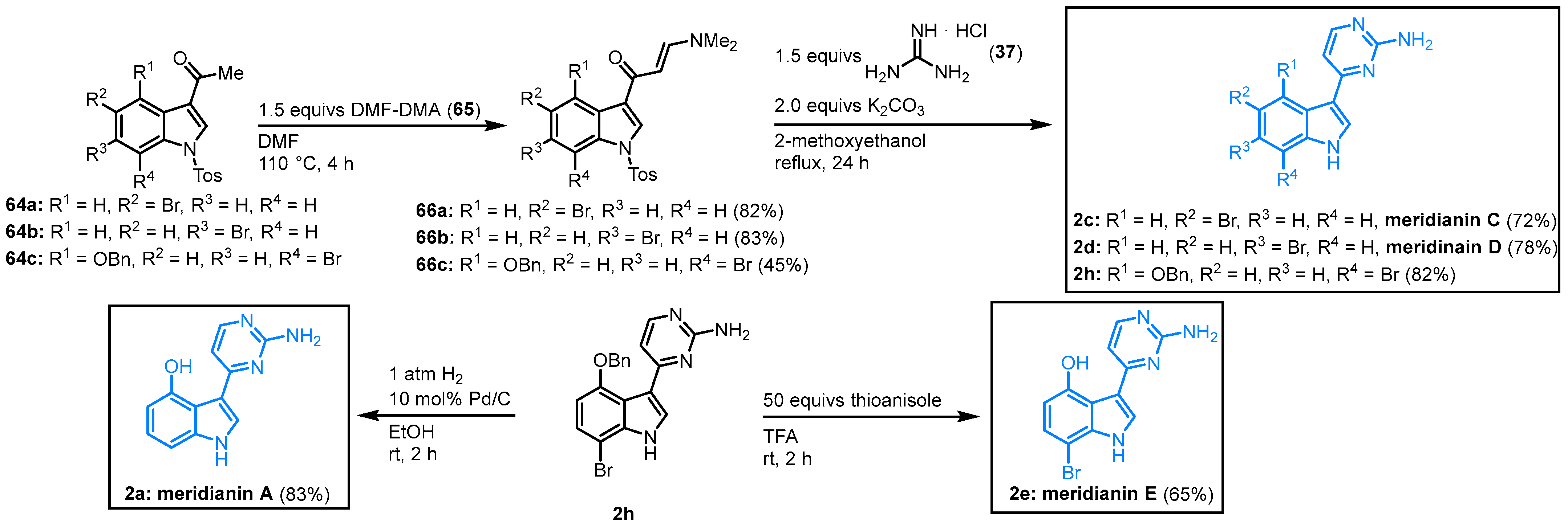
Shortly after the publication of the first meridianin syntheses, Fresneda and Molina developed a facile two-step synthesis of meridianins, starting from
N-tosyl-3-acetylindoles
64. The reaction of
64 with dimethylformamide dimethylacetal (DMF-DMA) gave the enaminone
66. The key step was the formation of the 2-aminopyrimidine ring by condensation of
66 with guanidine hydrochloride (
37). Molina and Fresneda described the synthesis of meridianin D (
2d, 65% overall yield) as well as the first total synthesis of meridianin C (
2c, 59% overall yield) and
O-benzyl-protected derivative
2h. After
O-deprotection and dehalogenation of
2h with hydrogen and palladium on carbon, meridianin A was synthesized for the first time (
2a, 31% overall yield) or respectively by treating
2h with the milder deprotecting agent; no dehalogenation occurred to give the first total synthesis of meridianin E (
2e, 24% overall yield) (
Figure 11)
[14][15].
Figure 11. Synthesis of meridianins A, C, D and E by Molina and Fresneda
[14].
2.3. Meridianin Synthesis by Müller via Carbonylative Alkynylation
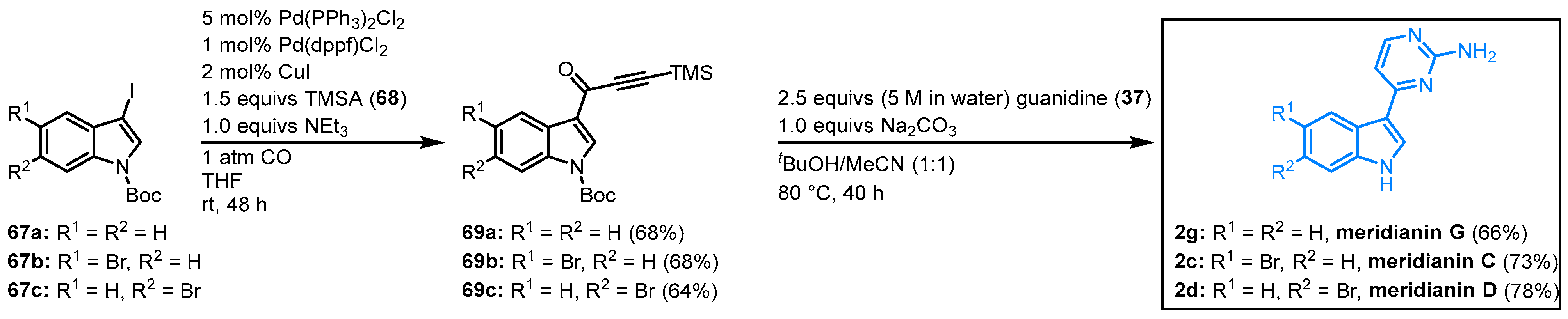
In 2005, Karpov et al. published a concise synthesis of meridianins C, D, and G. Tert-butoxycarbonyl(Boc)-protected indoles (67) reacted in a palladium-catalyzed three-component carbonylative alkynylation with TMS-protected acetylene (TMSA) (68) to the TMS-alkynones 69. Subsequent cyclocondensation with guanidine (37) concluded the meridianin synthesis as both the TMS- and the Boc-group are cleaved under the chosen reaction condition. Meridianin G was obtained with an overall yield of 45%, and meridianin C and D could be isolated with 50% overall yield (Figure 12).
Figure 12. Synthesis of meridianins C, D, and G by Karpov et al. via carbonylative alkynylation
[16].
2.4. Meridianin Synthesis by Penoni via Indolozation of Nitrosoarenes
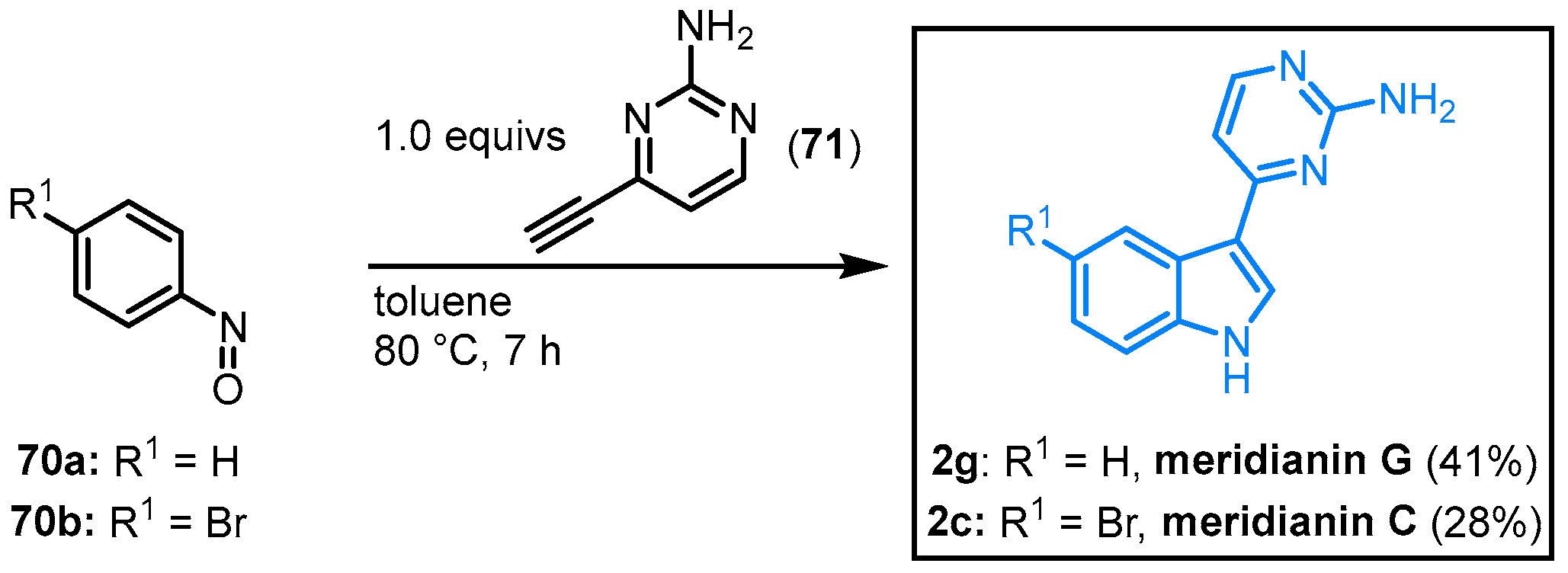
Efficiently, Penoni approached the synthesis of meridianins C and G. In a one-pot process, the corresponding nitrosobenzene
70 was reacted with 2-amino-4-ethynylpyrimidine (
71) to give the natural products
2c (28%) and
2g (41%) (
Figure 13)
[17].
Figure 13. Indolization of nitrosoarenes for the synthesis of meridianins C and G by Penoni
[17].
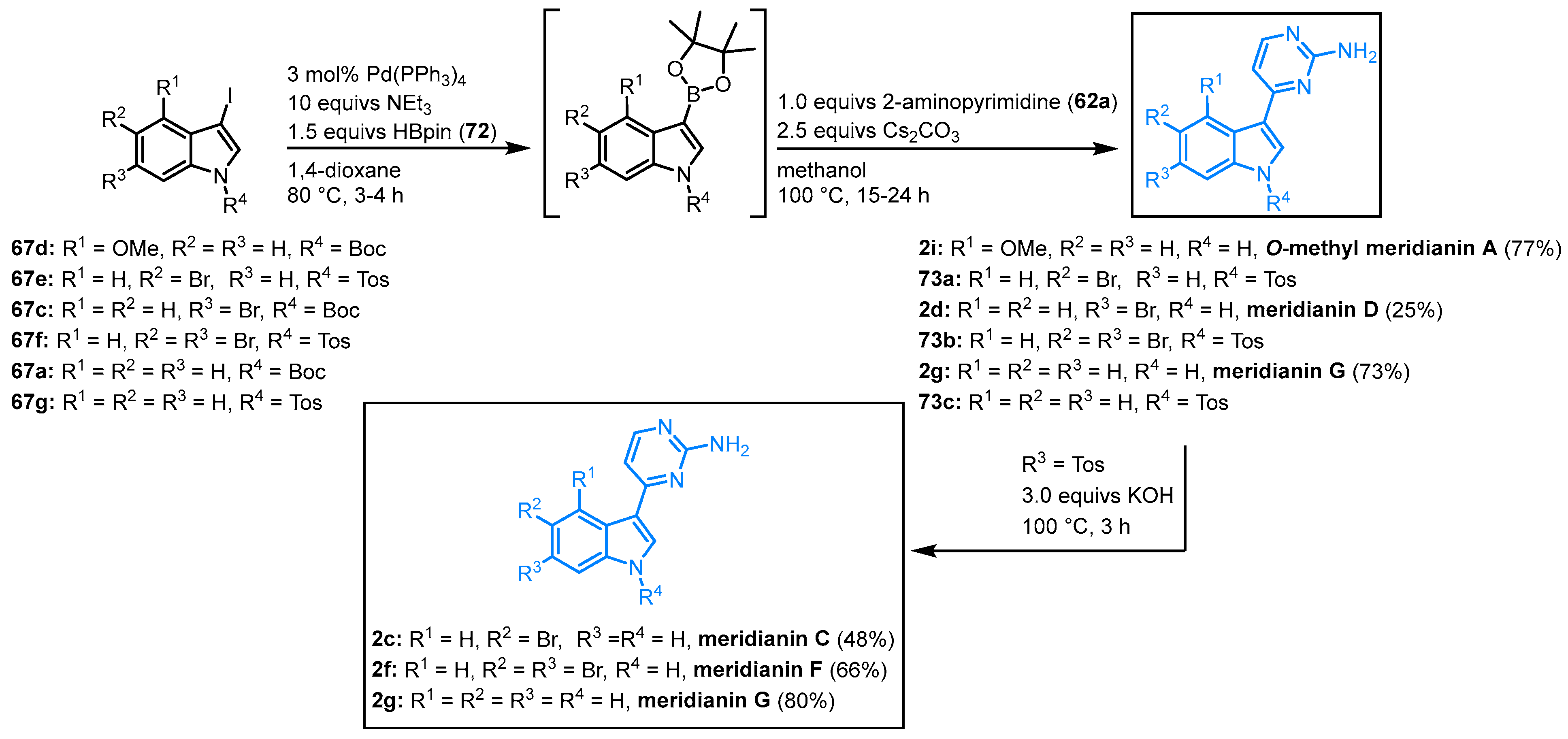
2.5. Synthesis of Meridianins via One-Pot Masuda Borylation-Suzuki Coupling Sequence by Müller
In 2011 and 2022, Müller and coworkers published a different synthetic strategy addressing meridianins. In a palladium-catalyzed Masuda borylation-Suzuki coupling (MBSC), one-pot procedure meridianins C, D, F and G, as well as the meridianin A precursor O-methyl meridianin A (2i), could be synthesized. 3-Iodo-N-protected indoles 67 react with pinacolyl borane (HBpin) (72) and without the isolation of the resulting boronic acid ester, the subsequent Suzuki coupling with 2-aminopyridine (62a) leads to the formation of O-methyl meridianin A (2i) and meridianins D (2d) and G (2g). The Boc-protecting group is cleaved under the Suzuki conditions. In contrast to this, N-tosyl-protected indoles 73 require an additional deprotecting step that can be implemented in the one-pot process. Treatment with potassium hydroxide leads to meridianins C (2c), F (2f), and G (2g) (Figure 14).
Figure 14. Synthesis of meridianins by Müller via MBSC sequence
[18][19].
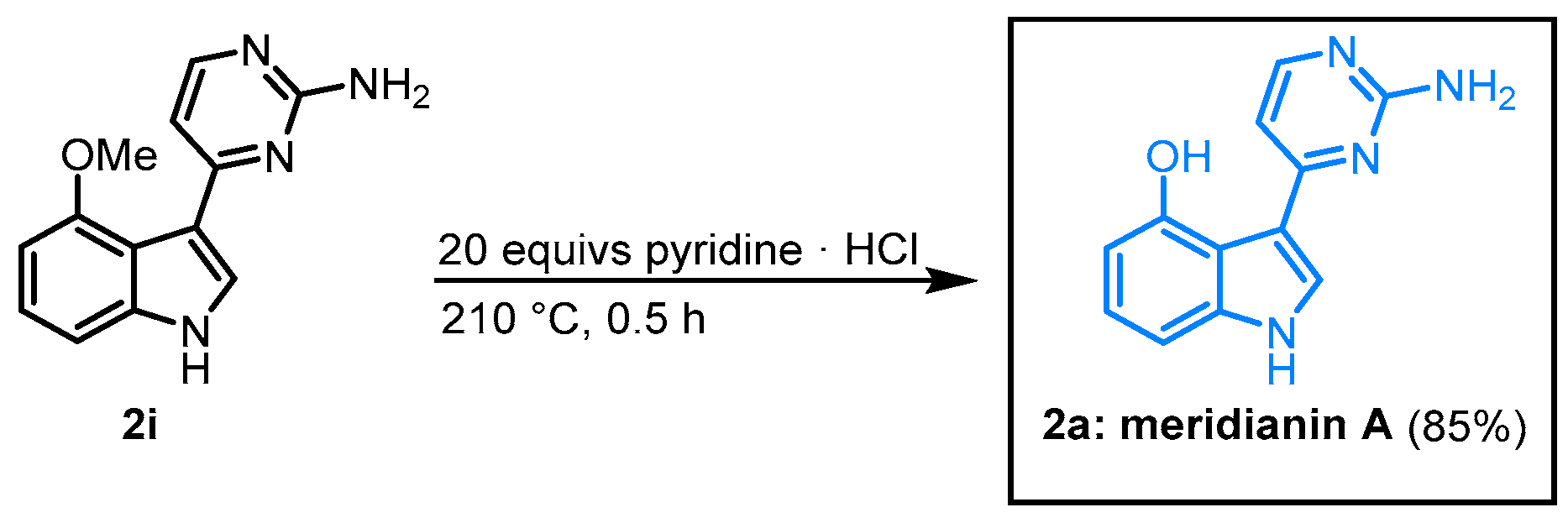
When
O-methyl meridianin A (
2i) was melted with pyridinium hydrochloride, the natural product
2a could be isolated in 85% (
Figure 15)
[18].
Figure 15. Demethylation of 2i furnished meridianin A.
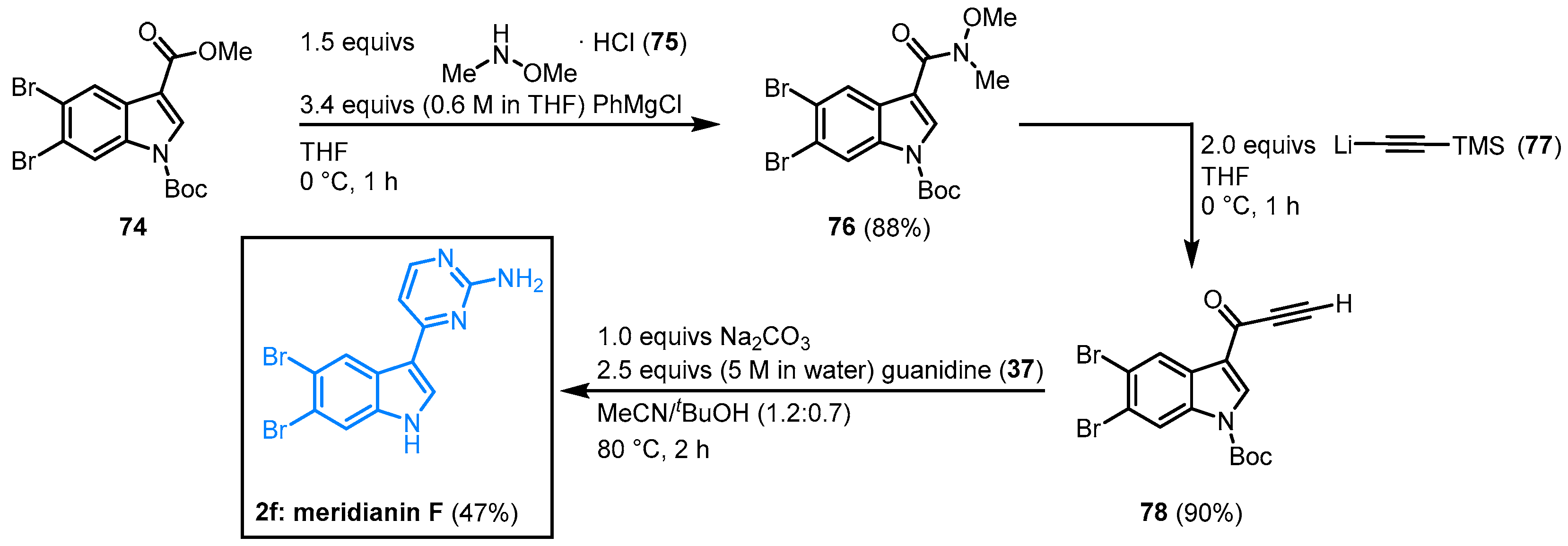
2.6. Synthesis of Meridianin F by Grainger
Grainger and coworkers worked on the regioselective dibromination of methyl indole-3-carboxylate and its application in the synthesis of indole building blocks. In this context, the synthesis of meridianin F was performed. Dibrominated indole
74 was reacted with
N,
O-dimethylhydroxylamine (
75) to form the corresponding Weinreb amide
76. Treatment with lithium(trimethylsilyl)acetylide (
77) led to the formation of alkynone
78. In the aftermath, meridianin F (
2f) was furnished by cyclocondensation with guanidine (
37) according to the aforementioned protocol by Müller in an overall yield of 37% (
Figure 16)
[16][20].
Figure 16. Synthesis of meridianin F by Grainger
[20].
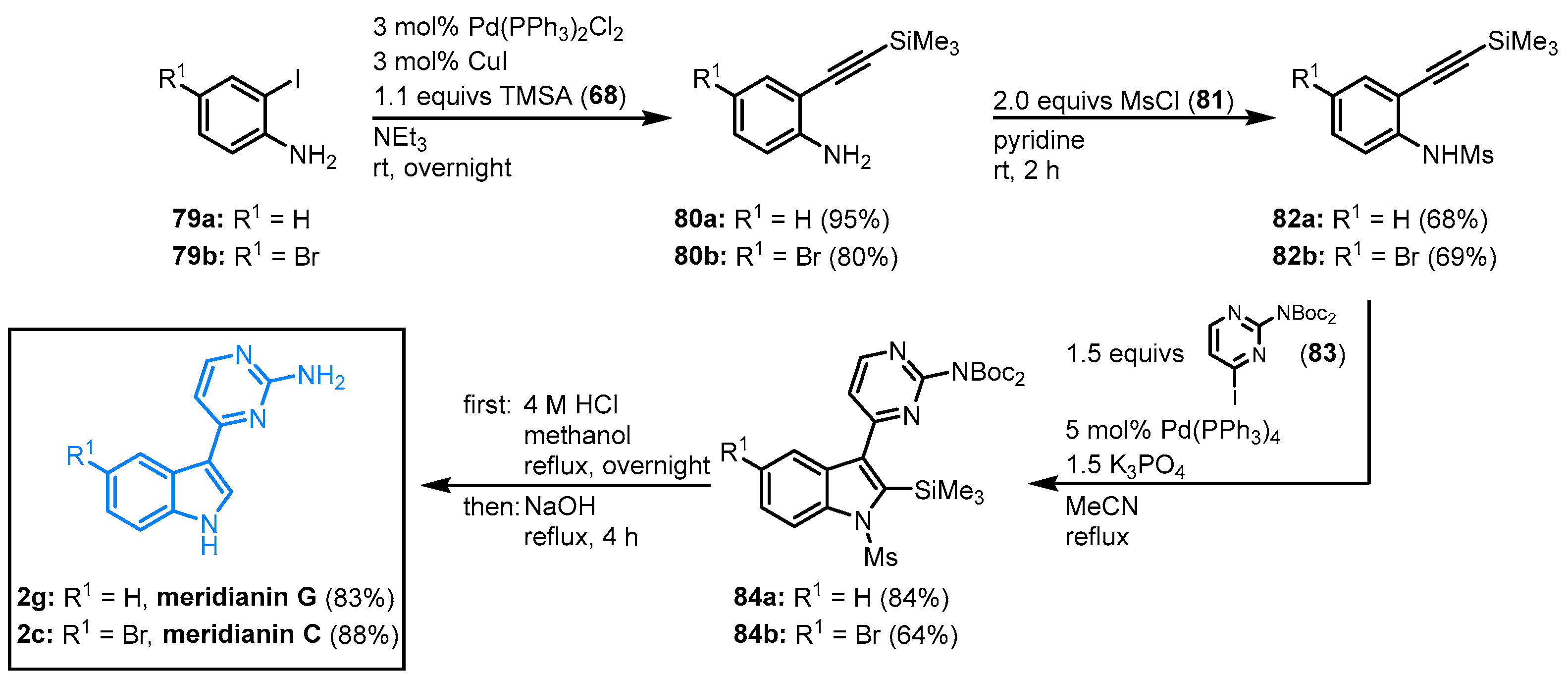
2.7. Domino Amino-Palladation Reaction for the Synthesis of Meridianins C and G by Morris
Morris and coworkers came up with a modified Cacchi protocol
[21] to synthesize meridianins from readily available monocyclic precursors in a catalytic domino amino-palladation reaction
[22]. The four-step synthesis starts with a Sonogashira coupling of 2-iodoaniline
79 and TMSA (
68) to give 2-alkynyl anilines
80 followed by
N-mesylation to give the activated sulfonamide
82. The reaction of
82 with the
N-Boc-protected 4-iodo-2-aminopyrimidine
83 in a Cacchi-type protocol led to the formation of the protected meridianin precursors
84. The global deprotection was achieved in a one-pot acid/base process and furnished meridianins C (
2c, 31% overall yield) and G (
2g, 45% overall yield) in four steps (
Figure 17).
Figure 17. Synthesis of meridianins C and G via palladium-catalyzed domino reaction by Morris
[22].
3. Syntheses of Meriolins
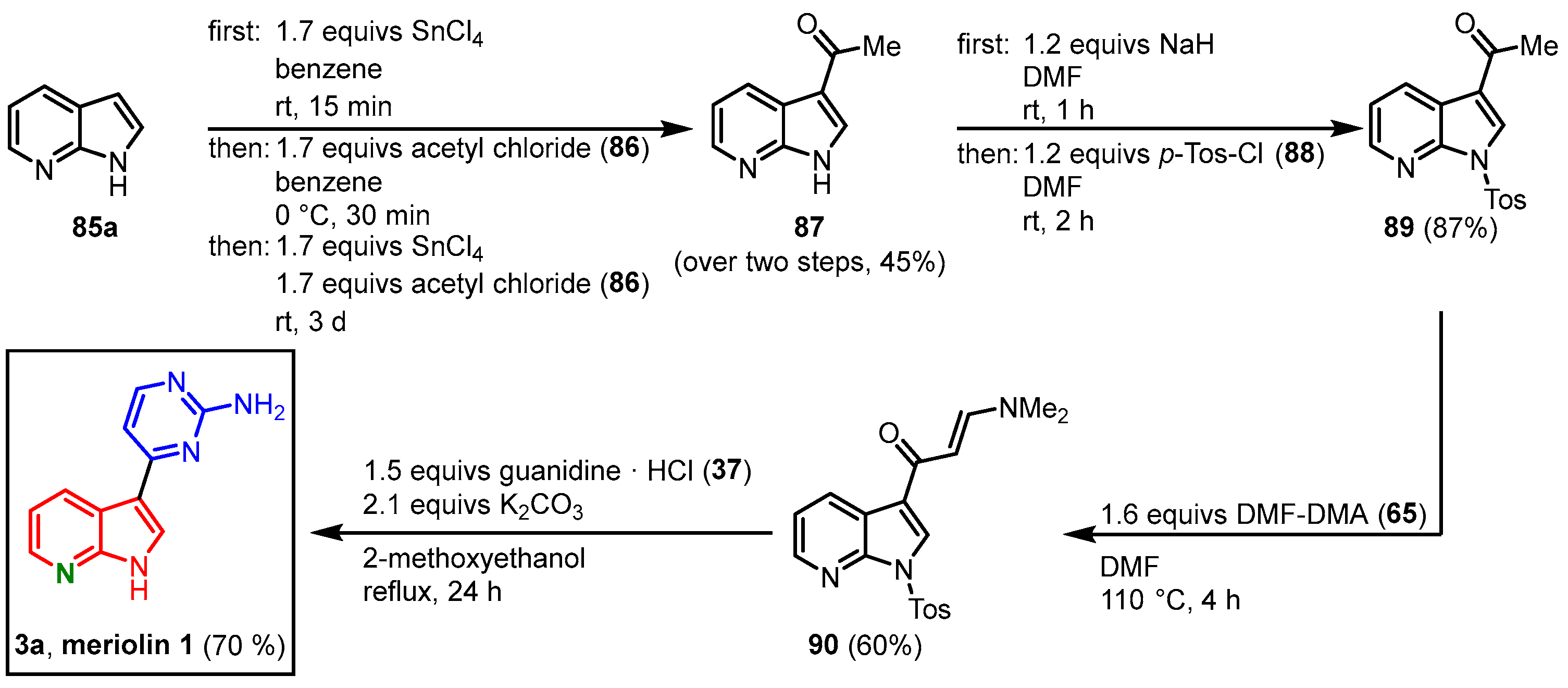
3.1. First Synthesis of Meriolin 1 by Molina and Fresneda
With their protocol for the synthesis of meridianins in hand, Molina and Fresneda were able to extend their strategy to 7-azaindoles, leading to the first synthesis of meriolin 1 (
3a). 7-Azaindole (
85a) was treated with acetyl chloride (
86) in the presence of tin (IV) tetrachloride, which afforded 3-acetyl-7-azaindole (
87). After
N-tosyl-protection, the enaminone
90 was furnished after the reaction of
89 with DMF-DMA (
65) similarly to the meridianin synthesis. Cyclocondensation with guanidine (
37) led to the 2-aminopyrimidine formation and meriolin 1 (
3a) was obtained in the 16% overall yield (
Figure 18)
[14].
Figure 18. Synthesis of meriolin 1 by Molina and Fresneda
[14].
3.2. Synthesis of Meriolin Derivatives by Joseph and Meijer
Joseph, Meijer, and coworkers used the strategy by Molina and Fresneda for the synthesis of meriolin 1 and in addition to that were able to generate a large substance library of novel meriolin derivatives. Starting from substituted 7-azaindoles
85, acylation in 3-position was achieved by treatment with acetic anhydride (
91) and trifluoroacetic acid.
N-protection with benzenesulfonyl chloride (
91) afforded the derivatives
94 (
Figure 19)
[23][24].
Figure 19. Preparation of 3-acetyl-
N-protected intermediates
94 [23].
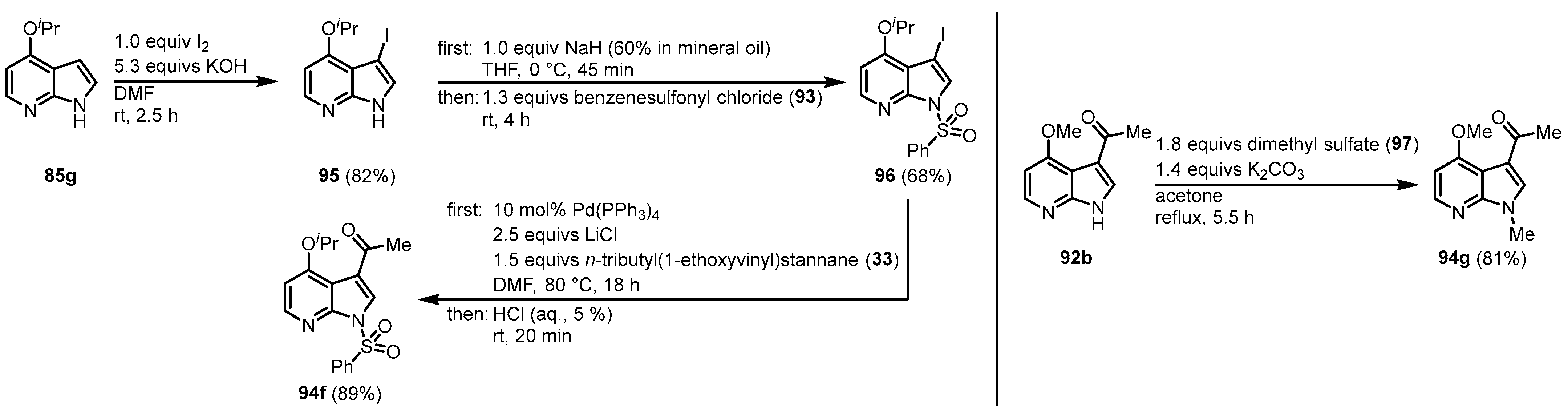
In the case of 85g, an alternative pathway was chosen to prevent O-demethylation under acidic conditions. After its iodination, the resulting 95 was first treated with benzenesulfonyl chloride (93) to give the N-protected intermediate 96 that was reacted with 33 in a palladium-mediated Stille cross-coupling reaction to form 94f. Treatment of the 4-methoxy derivative 92b with dimethyl sulfate (97) gave the N-methylated intermediate 94 g (Figure 20).
Figure 20. Alternative pathways to access
94f and
94g [23].
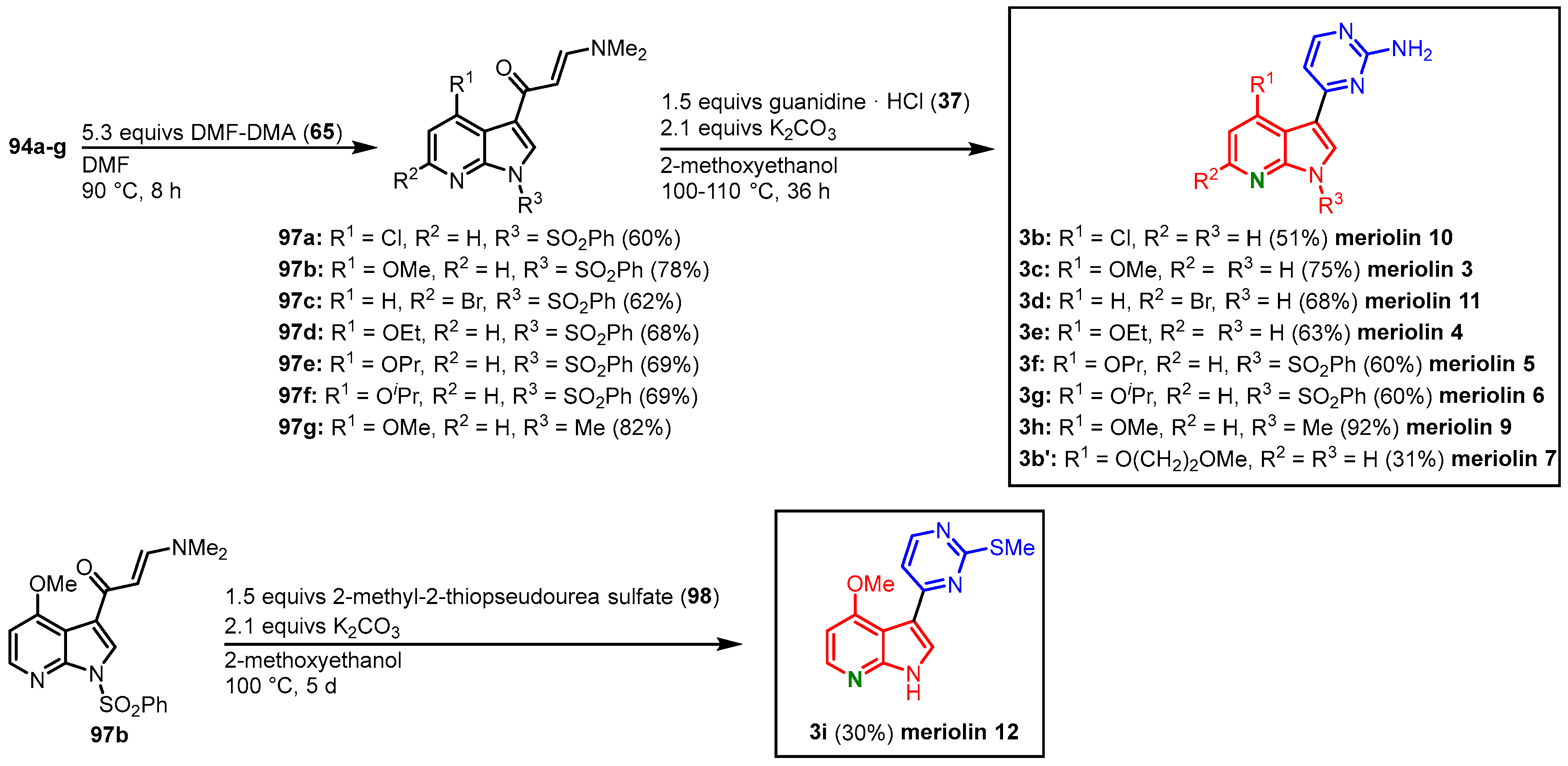
The functionalized 7-azaindoles 94 were then transformed to the corresponding enaminones 97 according to the Molina and Fresneda protocol, and after cyclocondensation with guanidine (37), meriolins 3–7 (3c, 3e–g, 3b’) and 9–11 (3h, 3b, 3d) were obtained. Meriolin 7 (3b’) was isolated as a side product in the synthesis of meriolin 10 (3b), where a nucleophilic substitution of the chlorine substituent took place. Treatment of 97b with 2-methyl-2-thiopseudourea sulfate (98) led to the formation of the 2-methylthiopyrimidine-substituted meriolin 12 (3i). The meriolins 3–7 and 9–12 were isolated in overall yields ranging from 12 to 37% starting from the corresponding 7-azaindole 85 (Figure 21).
Figure 21. Synthesis of meriolins 3–7 and 9–12 by Joseph and Meijer
[23].
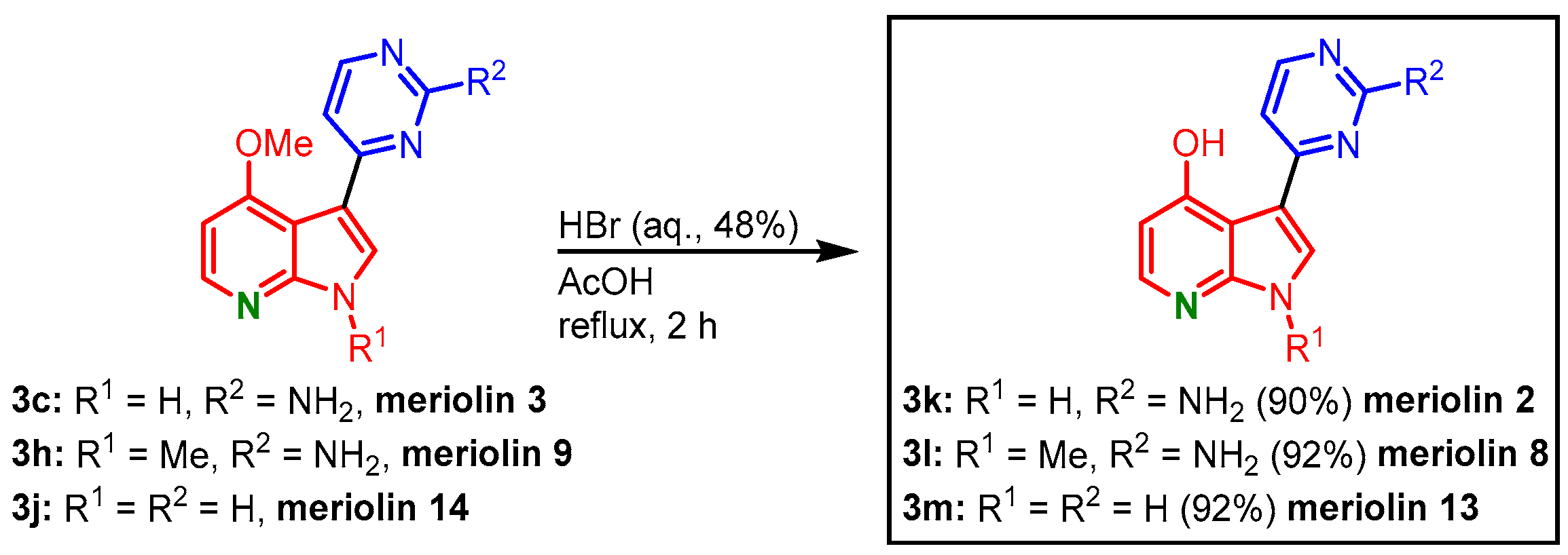
The 4-methoxy-substituted meriolins
3c,
3h, and
3j could be transformed to the corresponding 4-hydroxy-substituted meriolins 2 (
3k, 26% overall yield), 8 (
3l, 31% overall yield), and 13 (
3m, 22% overall yield) by
O-demethylation with hydrobromic acid in acetic acid (
Figure 22)
[23][24].
Figure 22. O-demethylation of meriolins 3, 9, and 14 gives 4-hydroxy-substituted meriolins 2, 8, and 13
[23].
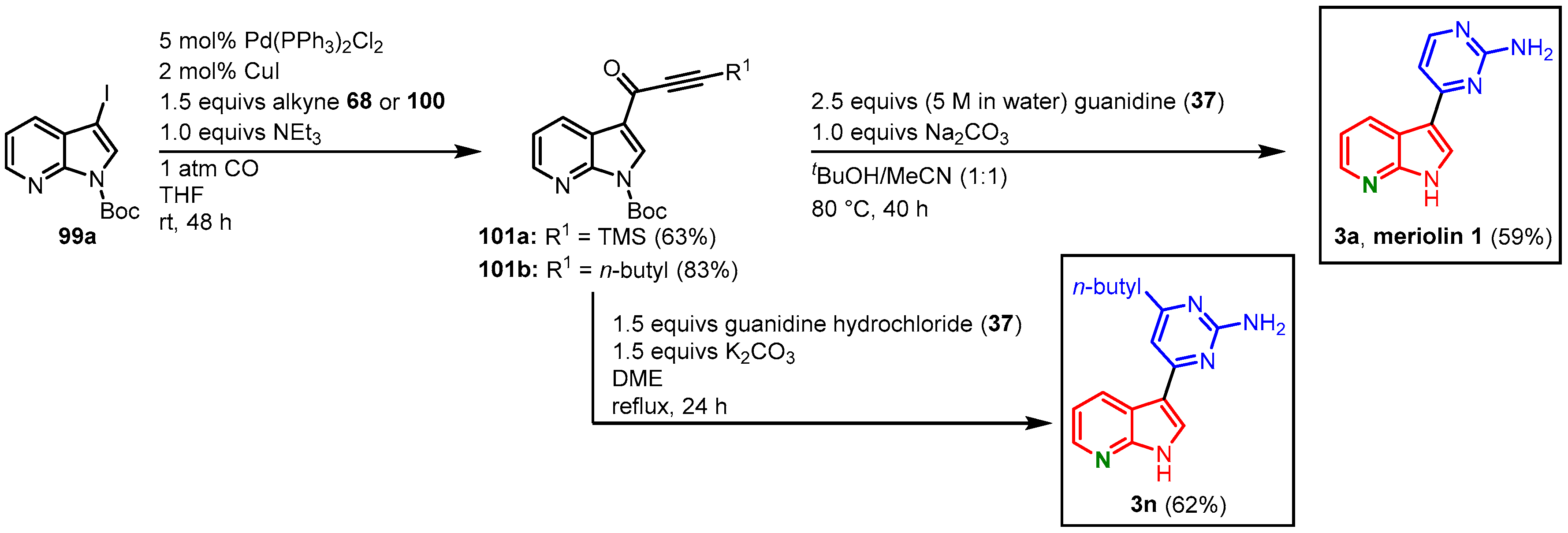
3.3. Meriolin Syntheses by Müller via Carbonylative Alkynylation
The Müller approach to meridianins via carbonylative alkynylation and subsequent pyrimidine synthesis could be transferred to the synthesis of meriolins. A small library of potential kinase inhibitors has been synthesized for screenings, among them meriolin derivatives
3a and
3n. Therefore, 3-iodo-
N-Boc-7-azaindole (
99a) was transformed to the alkynones
101 in a Sonogashira coupling with TMSA (
68) or 1-hexyne (
100). Alkynones
101 were then cyclized with guanidine (
37), either in a mixture of
tert-butanol and acetonitrile or in DMF, to the meriolin derivatives
3a (37% overall yield) and
3n (51% overall yield) (
Figure 23)
[16][25].
Figure 23. Synthesis of meriolins
3a and
3n via carbonylative alkynylation and subsequent pyrimidine synthesis by Müller
[16][25].
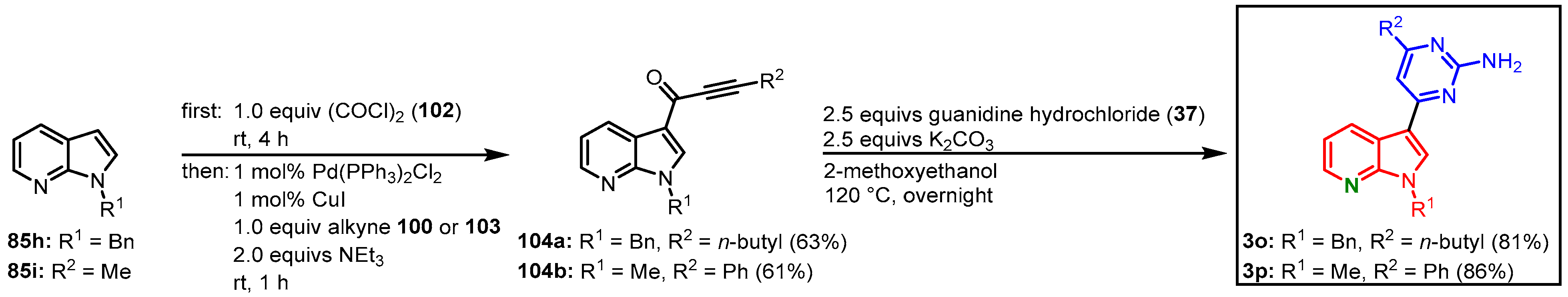
3.4. Three-Component Glyoxylation Decarbonylative Alkynylation Synthesis of Alkynones by Müller
Another approach by Merkul et al. to address meriolins was performed via a three-component glyoxylation alkynylation reaction, leading to
N-benzylated and
N-methylated meriolins
3o and
3p. Starting from 7-azaindoles,
85 in the first step reaction with oxalyl chloride (
102) furnished the indole-3-glyoxyl chlorides. These reactive synthetic equivalents of acid chlorides were directly transformed to alkynones
104 in a decarbonylative Sonogashira coupling with 1-hexyne (
100) or phenylacetylene (
103). The cyclocondensation reaction with guanidine (
37) went similarly as in the previously described strategy, which gave meriolins
3o and
3p in 51 and 52% overall yield (
Figure 24)
[26].
Figure 24. Synthesis of meriolins via three-component glyoxylation decarbonylative alkynylation by Merkul et al.
[26].
3.5. Synthesis of Meriolins with a Suzuki Coupling as a Key Reaction by Huang
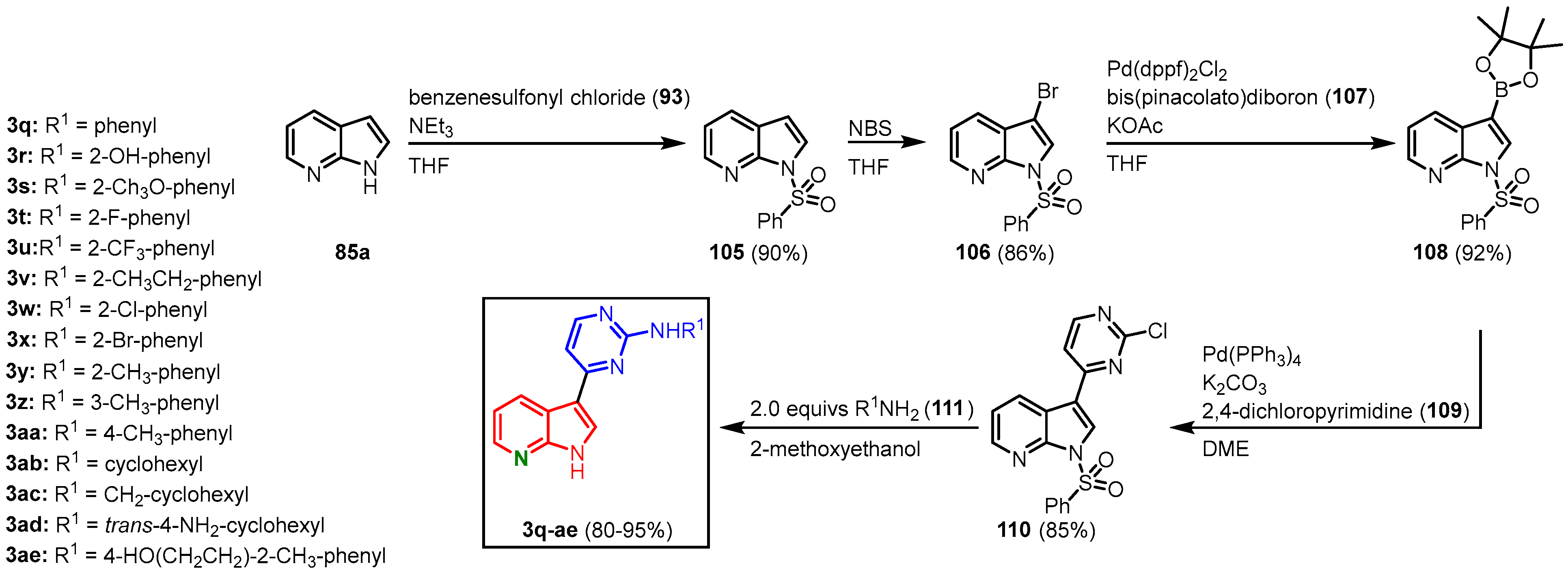
To investigate their kinase inhibitory effects, Huang and coworkers established a synthetic route to derivatize meriolins via a nucleophilic substitution on the pyrimidine moiety, as well as by functionalizing the N-1 and C-2 position on the azaindole moiety. 7-Azaindole (
85a) was
N-protected by treatment with benzenesulfonyl chloride (
93) before it was selectively brominated in the C-3 position. Brominated and
N-protected
106 was then transformed to the boronic acid ester
108 in a Pd(dppf)
2Cl
2-catalyzed Miyaura borylation with bis(pinacolato)diboron (
107). Suzuki coupling with 2,4-dichloropyrimidine (
109) in the presence of Pd(PPh
3)
4 gave
110 in superior regioselectivity. The chlorine substituent on the pyrimidine ring could then be substituted by various amines
111 in a nucleophilic aromatic substitution. Two equivalents of amine were used, as one equivalent was consumed by the concurrent cleavage of the benzenesulfonyl group. This furnished 15 meriolin derivatives (
3q-ae) with overall yields ranging from 48 to 58% (
Figure 25)
[27].
Figure 25. Meriolin synthesis by Huang via nucleophilic substitution on the pyrimidine moiety
[27].
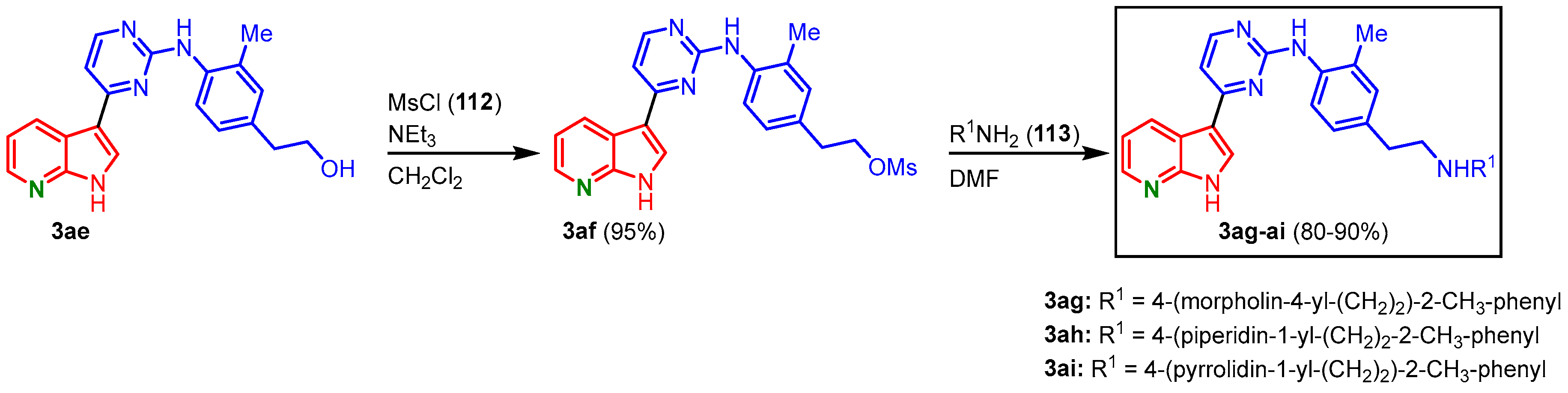
For the installment of solubilizing amino side chains, derivative
3ae was treated with methanesulfonyl chloride (
112), and the mesylate
3af was obtained. After treatment with different amines
113, the meriolin derivatives
3ag-
ai have been isolated in overall yields of 76–86% (starting from
3ae) (
Figure 26)
[27].
Figure 26. Introduction of solubilizing side chains gave the meriolin derivatives
3ag-
ai [27].
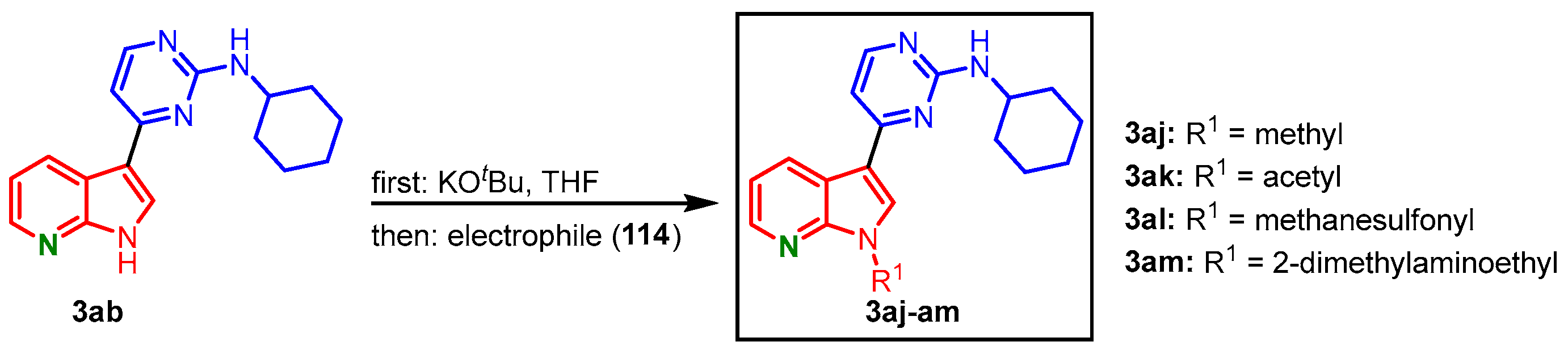
To assess the role of the NH group of the 7-azaindole unit in CDK1 binding,
N-functionalization was anticipated. Compound
3ab was treated with potassium
tert-butoxide before it reacted with different electrophiles
114 to give the derivatives
3aj-
am (
Figure 27)
[27].
Figure 27. N-functionalization of compound
3ab with different electrophiles gave meriolins
3aj-
am [27].
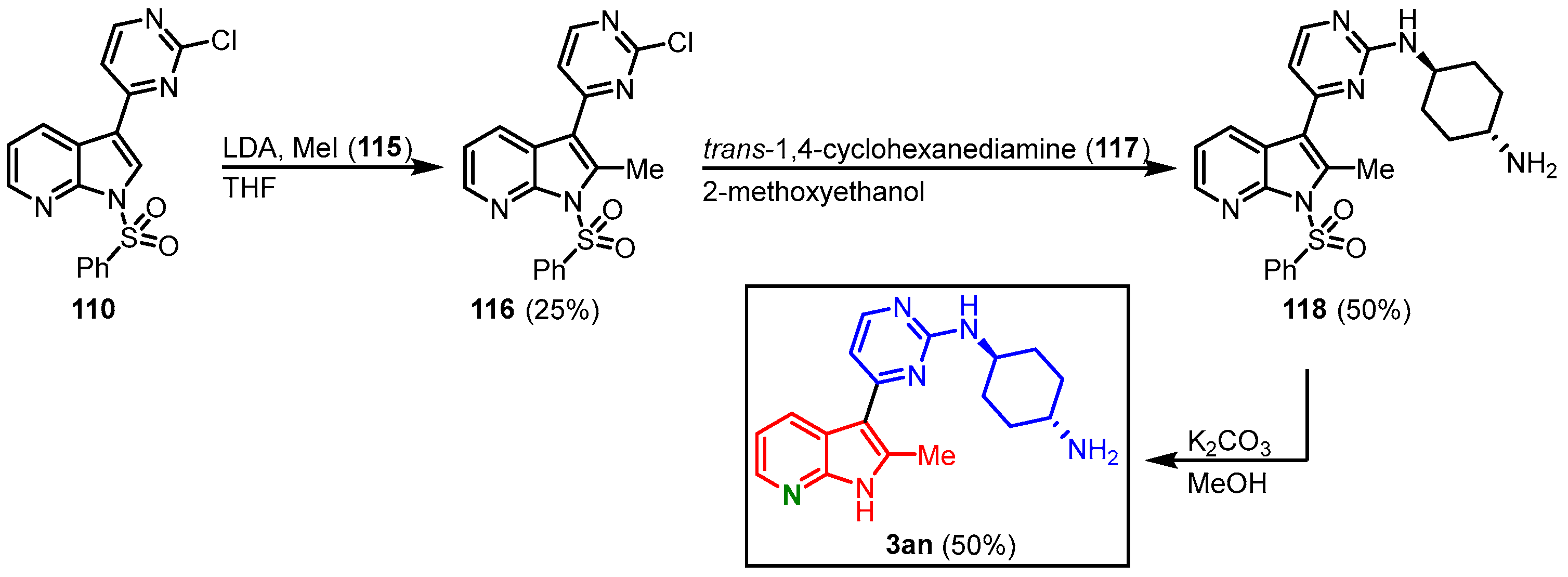
Lastly, compound
110 was treated with lithiumdiisopropylamine (LDA) and methyl iodide (
115) to introduce a methyl group in the C-2 position. After the reaction with amine
117, an additional deprotection step was added, since the C-2 methyl group caused the
N-benzenesulfonyl group to be stable under hot aminolysis conditions. This furnished meriolin
3an in 6% overall yield starting from
110 (
Figure 28)
[27].
Figure 28. Synthesis of meriolin
3an [27].
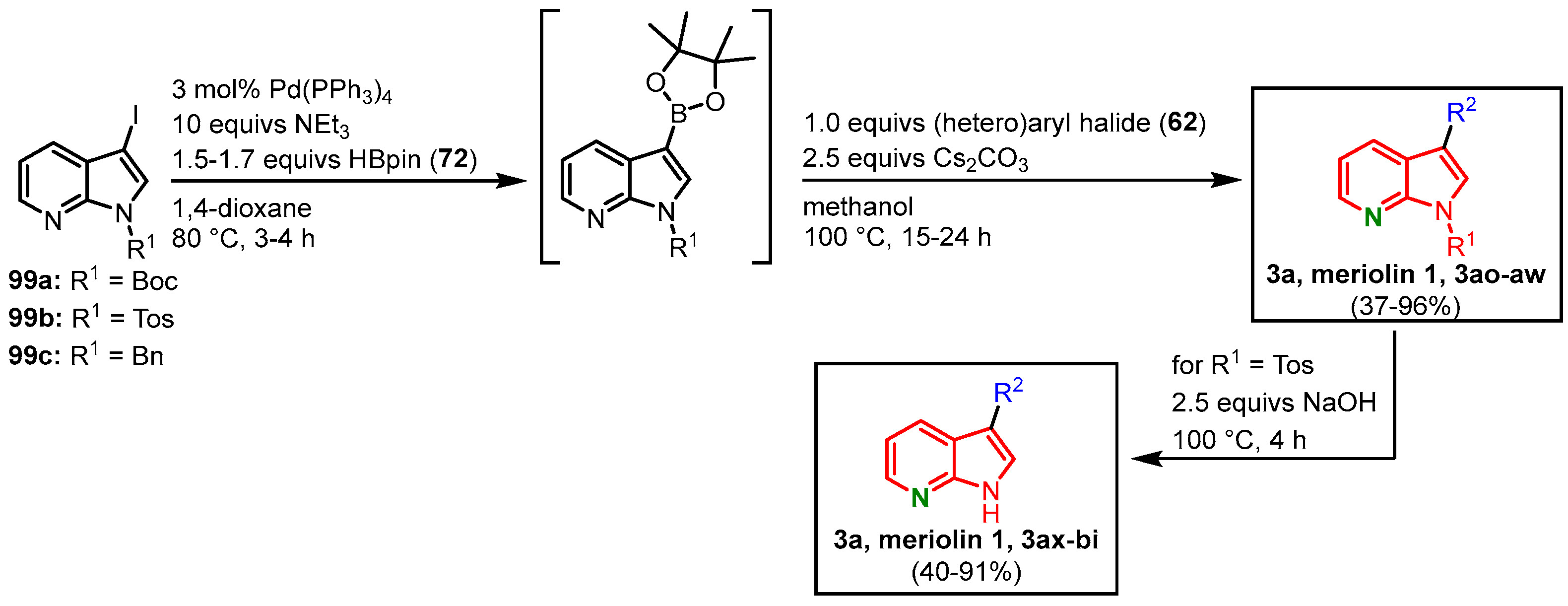
3.6. Meriolin Synthesis via the Masuda borylation-Suzuki Coupling Sequence by Müller
The Müller group could show the versatility of the MBSC sequence by transferring their meridianin protocol to the synthesis of meriolins and other biaryl systems
[18][19][28][29]. In a one-pot-process, 7-azaindoles
99 were transformed to the corresponding pinacolyl boronic acid esters in a palladium-mediated Masuda borylation. In the sense of a sequentially catalyzed reaction, a subsequent Suzuki coupling with arylhalide
62 follows (
Figure 29). Under Suzuki conditions, the Boc group is concomitantly cleaved, leading to meriolin 1 (
3a), meriolins
3ao-
au, and the
N-benzylated meriolins
3av and
3aw, with yields ranging from 37 to 96% (
Table 1). If the reaction sequence is started with
N-tosylated azaindoles
99b a subsequent deprotection step with a hydroxide base is required. This step can be included in the one-pot process, which led to meriolins
3a and
3ax-
bi with overall yields ranging from 40 to 91% starting from 7-azaindoles
99b (
Figure 29)
[18][28].
Figure 29. MBSC sequence for the synthesis of meriolins
3a and
3ao-
bi by Müller
[18][28].
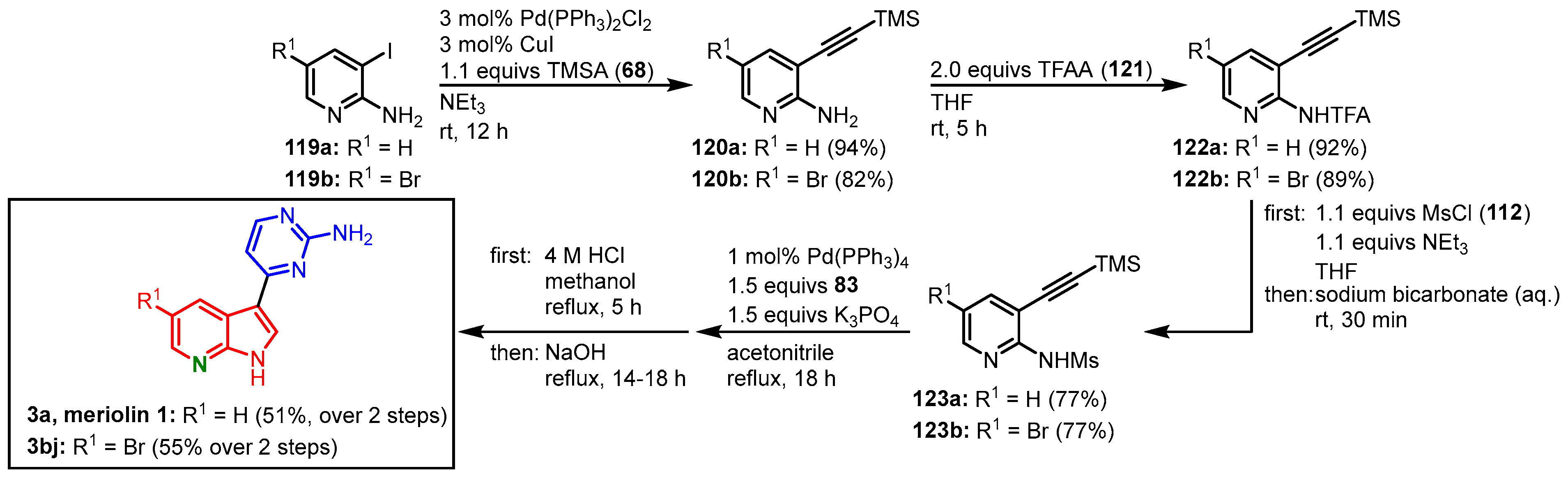
Morris and coworkers tried to adapt their meridianin protocol to the synthesis of meriolins starting from iodinated aminopyridines
119 to give 3-alkynylated 2-amino pyridines
120. In contrast to anilines (vide supra), it was not possible to prepare the monomesylated aminopyridines directly, which required treatment with trifluoroacetyl anhydride (TFAA) (
121) first to furnish trifluoroacetamides
122. Reaction with mesyl chloride (
112) led to the desired intermediate
123 that could be converted in the optimized domino reaction with
N-Boc-4-iodopyrimidine-2-amine (
83) and subsequent acid/base deprotection protocol to give meriolin 1 (
3a) in 34% overall yield, as well as the 5-bromo meriolin
3bj in 31% overall yield (
Figure 30)
[22].
Figure 30. Synthesis of meriolins
3a and
3bj via domino amino-palladation reaction by Morris
[22].
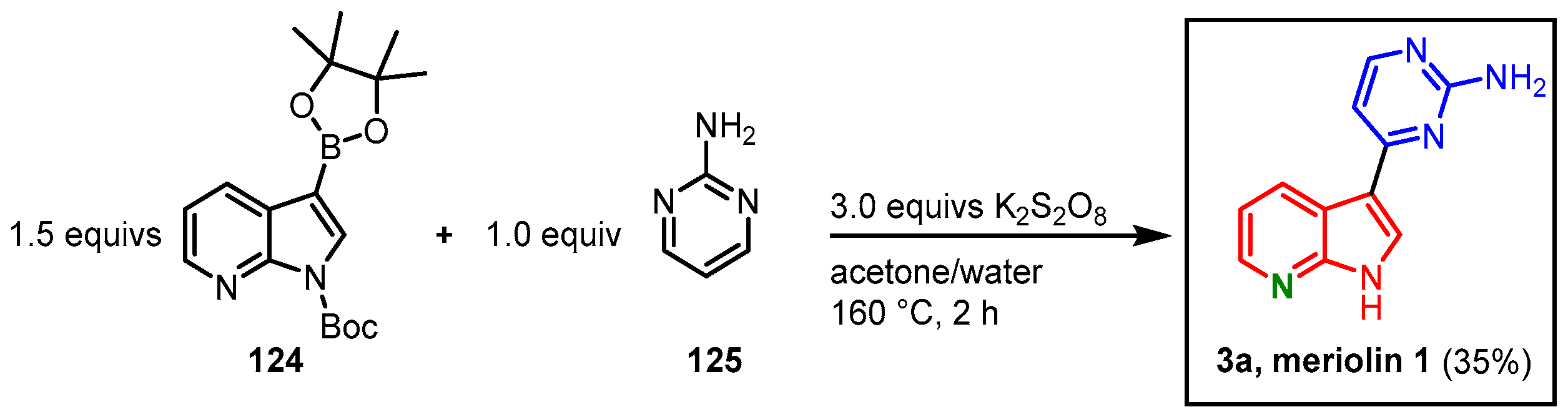
3.8. Metal-Free CH-Activation of a Pyrimidine and an Indolylboronic Ester by Singh
In 2016, Singh presented a metal-free CH-activation approach toward the synthesis of meriolin 1. The group reported cross-coupling between diazines and related electron-deficient heteroarenes with organoboron species. Treatment of
N-Boc-protected boronic acid ester
124 with 2-aminopyrimidine (
125) and potassium persulfate in an acetone-water mixture led to the formation of meriolin 1 in 35% yield (
Figure 31). The proposed mechanism includes the formation of a sulfate anion radical that activates the boronic acid ester and generates an azaindolyl radical. The radical reacts with the in situ-formed pyrimidinyl salt to form a radical cation. After it undergoes single electron transfer, the protonated form of the desired product is obtained
[30].
Figure 31. Metal-free synthesis of meriolin 1 via CH-activation by Singh
[30].
3.9. Functionalization of Meriolins via Suzuki Coupling or Nucleophilic Substitution Reactions by Singh and Malik
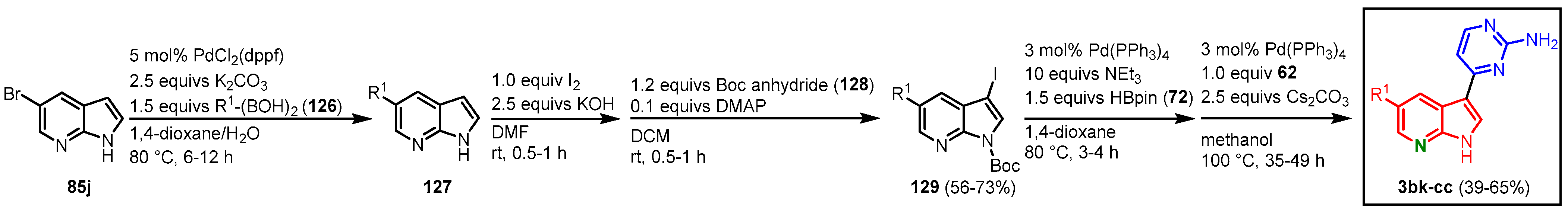
A different approach by Singh in cooperation with Malik was more pragmatic to synthesize a large library of meriolins to establish structure-activity-relationships and determine their potency against CDKs. It was elaborated that functionalization in the C-5 position and N-1 position on the 7-azaindole unit, as well as on the pyrimidine ring, should be accomplished. Starting from 5-bromo-7-azaindole (
85j) in a Suzuki coupling with several boronic acids,
126 led to functionalized 7-azaindoles
127 (
Figure 33). After iodination and protection with Boc-anhydride (
128) to give 3-iodo-7-azaindoles
129, a Masuda borylation with pinacolyl borane (
72) and subsequent Suzuki coupling with 4-chloropyrimidine-2-amine (
62) gave the meriolin derivatives
3bk-
cc in 25–46% overall yield (
Figure 33,
Table 2)
[31].
Figure 33. Functionalization of the C-5 position led to meriolins
3bk-
cc [31].
Table 2. Boronic acids 126 used for the functionalization in C-5 position and the corresponding yields of meriolins 3bk-cc.
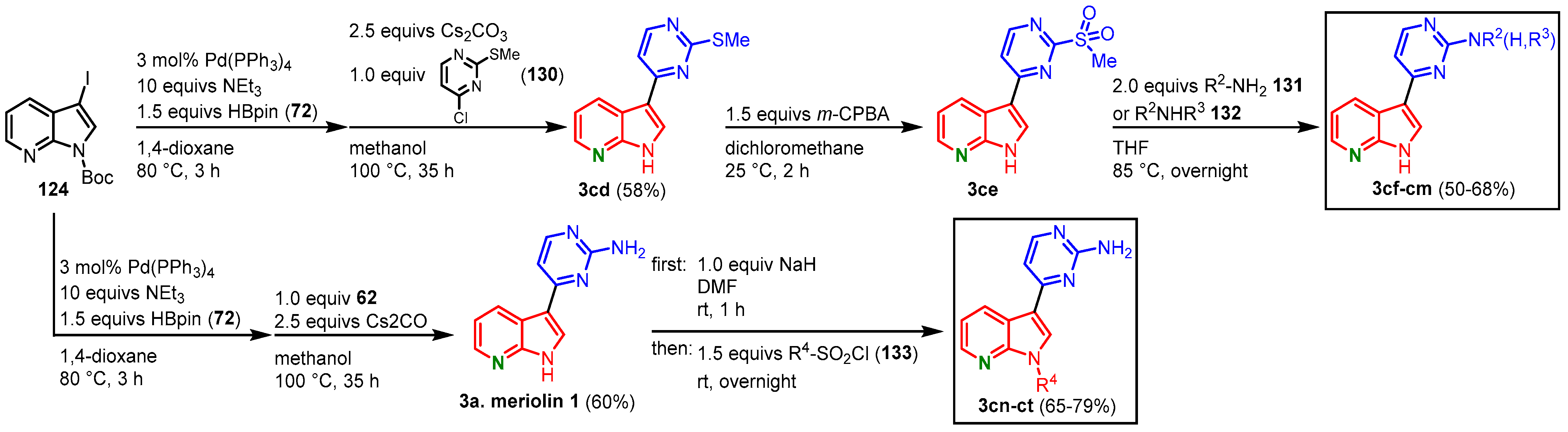
To derivatize the pyrimidine ring, iodinated and N-protected 7-azaindole 124 was transformed to the corresponding pinacolyl boronic ester and reacted with 130 in a Suzuki coupling to give compound 3cd. The thiomethyl group was oxidized to give the sulfone 3ce using m-CPBA. Nucleophilic substitution by several primary 131 or secondary amines 132 furnished meriolins 3bk-cc in 29–39% overall yield (Figure 33, Table 3). To vary the substituents in the N-1 position, at first the synthesis of meriolin 1 was approached using a Masuda borylation and subsequent Suzuki coupling. After treatment with sodium hydride, reaction with different sulfonyl chlorides 133 gave meriolins 3cn-ct in 39–47% overall yield (Figure 33, Table 4).
Figure 33. Functionalization of the pyrimidine ring and the N-1 position gave meriolins
3cf-
ct [31].
Table 3. Primary 131 and secondary amines 132 used for the functionalization of the pyrimidine ring and the corresponding yields of meriolins 3cf-cm.
Table 4. Sulfonyl chlorides 133 for the functionalization in N-1 position and the corresponding yields of meriolins 3cn-ct.
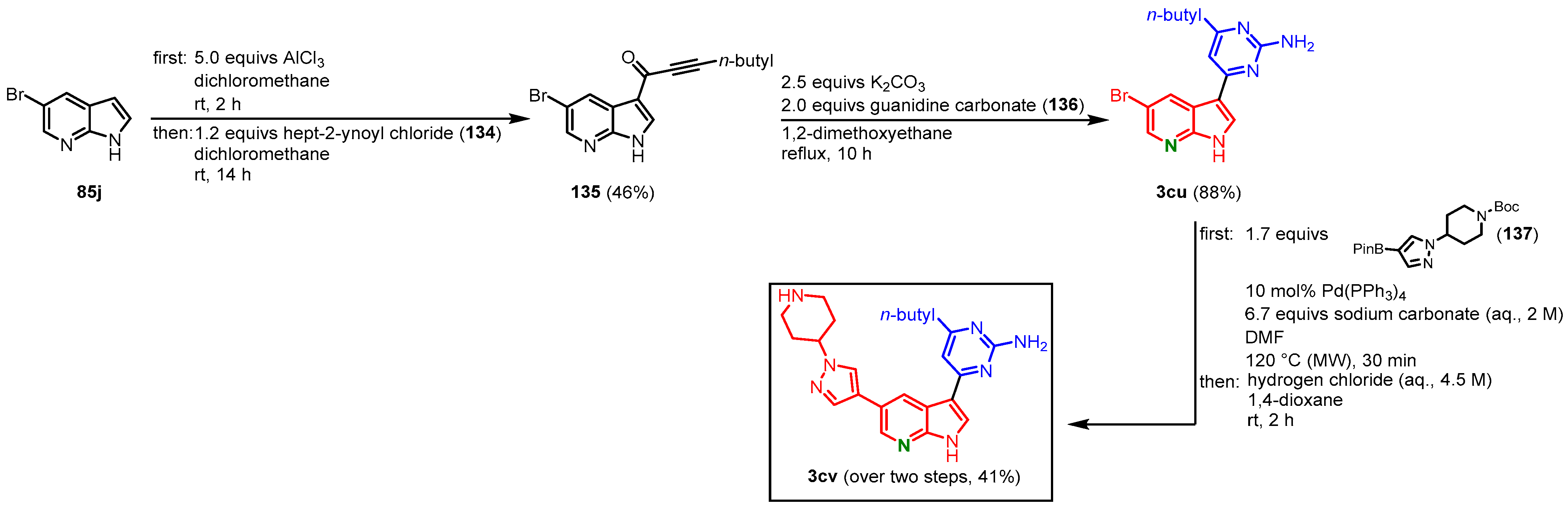
3.10. Meriolin Synthesis via Friedel Crafts Acylation by Grädler
Grädler and coauthors started their approach on meriolins from 5-bromo-7-azaindole (
85j) with a Friedel Crafts acylation using aluminium chloride and acid chloride
134. The intermediate
135 was reacted in a cyclocondensation with guanidine carbonate (
136), which furnished meriolin
3cu in 40% overall yield. The bromine atom in C-5 position was then employed for further derivatization. Suzuki coupling with Boc-protected pinacolyl boronic acid ester
137 and subsequent Boc-deprotection with hydrochloric acid gave meriolin
3cv in 41% yield (
Figure 34). The overall yield after three steps is 17%
[25]. Using this method, as well as the carbonylative alkynylation by Müller
[16], several derivatives have been synthesized and tested for their PDK1 inhibitory properties
[25].
Figure 34. Preparation of meriolin
3cv via Friedel Crafts acylation by Grädler
[25].