1. Introduction
The 1,3-dipolar cycloaddition reaction of nitrile oxides as 1,3-dipoles and alkenes/alkynes as dipolarophiles has become an efficient tool in organic synthesis to obtain various substituted isoxazolines/isoxazoles [1,2,3]. The reaction was developed by Rolf Huisgen and described by Albert Padwa in their investigations on 1,3-dipolar cycloadditions [4,5]. Nitrile oxides, which are typically generated in situ, undergo subsequent 1,3-dipolar cycloaddition to form appropriate isoxazoles or isoxazolines. Numerous methods of nitrile oxide generation have been reported, mainly including the dehydration of nitroalkanes [6,7,8] and oxidation of aldoximes [9,10,11]. Alternatively, Svejstrupor described the synthesis of isoxazolines and isoxazoles from hydroxyimino acids via the visible-light-mediated generation of nitrile oxides by two sequential oxidative single electron transfer processes [12]. More recently, Chen et al. reported the synthesis of fully substituted isoxazoles from nitrile oxides, which were generated in situ from copper carbene and tert-butyl nitrite [13].
Notably, the intramolecular nitrile oxide cycloaddition (INOC) reaction can provide a route for the preparation of isoxazoles or isoxazolines annulated to various carbo- or heterocycles. For example, the intramolecular 1,3-dipolar cycloaddition of 2-phenoxybenzonitrile N-oxides to neighboring benzene rings, accompanied by dearomatization, formed the corresponding isoxazolines in high yields [14]. Recently, a method for the stereoselective synthesis of novel isoxazoline/isoxazole-fused indolizidine-, pyrrolizidine- and quinolizidine-based iminosugars has been developed, employing N-alkenyl/alkynyl iminosugar C-nitromethyl glycosides as nitrile oxide precursors in 1,3-dipolar cycloaddition reactions [15]. The phthalate-tethered INOC strategy has also been described as a novel method for the synthesis of 12–15-membered chiral macrocycles having a bridged isoxazoline moiety in a highly regio- and diastereoselective manner [16]. Furthermore, diversity-oriented access to isoxazolino and isoxazolo benzazepines as possible bromodomain and extra-terminal motif protein (BET) inhibitors has been reported via a post-Ugi heteroannulation involving the intramolecular 1,3-dipolar cycloaddition reaction of nitrile oxides with alkenes and alkynes [17]. In addition, an intramolecular 1,3-dipolar nitrile oxide cycloaddition strategy has been applied as an efficient synthesis protocol for the regio- and diastereoselective construction of highly functionalized tricyclic tetrahydroisoxazoloquinolines [18].
Fused isoxazoles or isoxazolines obtained by the INOC reaction may also serve as synthetically important intermediates for many biologically active compounds. Such compounds, including the HBV inhibitor entecavir [19,20], the antibiotic branimycin [21], the antiviral (+)-Brefeldin A [22], tricyclic isoxazoles combining serotonin (5-HT) reuptake inhibition with α2-adrenoceptor blocking activity [23] and the alkaloids meliacarpinin B [24] and Palhinine A [25], have been synthesized by employing INOC as a key step.
2. Results
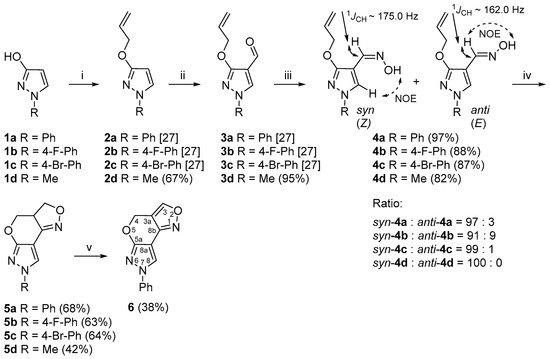
The synthetic strategy that we designed to construct the pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole ring system employs difunctional substrates (
4a–
d) that contain an aldoxime unit next to the allyloxy group attached to the pyrazole core and can serve as intermediates for nitrile oxide generation and subsequent cycloaddition (
Scheme 1).
Scheme 1. Synthetic route for the 3a,4-dihydro-3H,7H-pyrazolo[4′,3′:5,6]pyrano[4,3-c][1,2]oxazole ring system. Reagents and conditions: (i) NaH, DMF, 0 °C, 15 min, allylbromide, 60 °C, 1 h; (ii) DMF, POCl3, −10 °C, 15 min, 70 °C, 1 h; (iii) NH2OH·HCl, NaOAc, EtOH, reflux, 15 min; (iv) aq. NaOCl, DCM, rt, 1 h; (v) MnO2, toluene, reflux, 4 h.
As starting materials for the synthesis of compounds
4a–
d, we used 1-phenyl-, 1-(4-fluorophenyl)-, 1-(4-bromophenyl)- and 1-methylpyrazol-3-ols (
1a–
d), which are readily accessible from the oxidation of appropriate pyrazolidin-3-ones [
34]. The
O-allylation of
1a–
d with allylbromide in the presence of NaH gave
O-allylated pyrazoles
2a–
d [
27]. To introduce a formyl group to the 4-position of the pyrazole ring, we employed a previously reported Vilsmeier–Haack reaction procedure [
27,
35]. Heating compounds
2a–
d with Vilsmeier–Haack complex at 70 °C resulted in the formation of the desired pyrazole-4-carbaldehydes
3a–
d (
Scheme 1).
In order to prepare the 3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole derivatives
5a–
d by the INOC reaction, aldoximes
4a–
d were synthesized by the treatment of
3a–
d with hydroxylamine hydrochloride in the presence of sodium acetate [
36]. As a result, the
syn- and
anti-3-allyloxy-4-pyrazolaldoximes were obtained in total yields of 82–97%. The
1H NMR spectra of aldoximes
4a–
c showed the presence of two isomers in different ratios with a predominance of the
syn isomer, while the compound
4d was obtained as a pure
syn isomer.
Over the years, the isomerism of aldoximes has been thoroughly studied and many different NMR-based approaches have been developed, mainly due to the large differences in chemical shifts, coupling constants and distinct through-space connectivities in NOESY measurements of aldoxime
syn-
anti isomers [
37]. The configurational assignment of aldoximes
4a–
c was relatively easy due to the presence of both isomers, as it is well established that the resonance of the iminyl-H proton in the
syn isomer is greatly shifted upfield by approximately δ 0.5–0.7 ppm in the
1H NMR spectra compared to the
anti isomer [
38]. Moreover, the 1D selective NOESY experimental data of aldoximes
4a–
c showed that, upon irradiation of the hydroxyl proton N-OH of the predominant
syn isomer, a strong positive NOE on the pyrazole 5-H proton was observed, while the minor isomer showed a positive NOE on the iminyl-H, therefore confirming the
anti configuration. Finally, a heteronuclear 2D
J-resolved NMR experiment was used in order to determine
1JCH coupling constants throughout the series of aldoximes. It is well established from previous studies that there is a large and constant difference between the magnitudes of
1JCH coupling constants of the iminyl moiety in
syn-
anti isomers [
39], which is larger by at least 10–15 Hz for the
syn isomer. The measurements of compounds
4a–
c showed that the relevant
1JCH coupling constants of the iminyl moiety were around 175.0 Hz for the predominant
syn isomer, while the minor
anti isomer provided significantly lower coupling constant values by around 13.0 Hz. The configuration of aldoxime
4d as a pure
syn isomer was easily deduced from NOESY measurements and the
1JCH coupling constant of the iminyl moiety, which was 174.5 Hz. The analysis of
15N NMR spectroscopic data showed highly consistent chemical shift values within each isomer, in a range from δ −18.2 to −25.7 ppm in the case of the
syn isomer and in a range from δ −15.6 to −16.5 ppm for the
anti isomer. A comparison of the relevant NMR data of aldoximes is presented in
Table S1.
Several methods for the oxidation of aldoximes to nitrile oxides are known in the literature, including the application of oxidants such as chloramine T [
40,
41],
N-halosuccinimides (NXS) [
42,
43,
44], hypohalites [
45,
46,
47], hypervalent iodine reagents [
48,
49,
50] and oxone [
51,
52,
53,
54]. The reaction conditions for 3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole ring formation were optimized by using
4a as a model compound (
Table 1). When treating aldoxime
4a with chloramine T in EtOH at 50 °C for 30 min, the polycyclic product
5a was obtained in poor (20%) yield (
Table 1, Entry 1). The intramolecular cyclization reaction of
4a in the presence of the aq. NaOCl in DCM gave the desired product
5a in 1 h in sufficient (68%) yield (
Table 1, Entry 2). The experiment with TEA as an additive did not improve the yield of the product and
5a was obtained in 52% yield (
Table 1, Entry 3). A similar result showing that no additional base is required to facilitate the cycloaddition was also observed by Roy and De in their investigation on the rate enhancement of nitrile oxide cyclization and, hence, rapid synthesis of isoxazolines and isoxazoles [
55].
Table 1. Optimization of the INOC reaction conditions for 5a synthesis.
| Entry |
Conditions |
Yield *, % |
| 1 |
Chloramine T, EtOH, 50 °C, 30 min |
20 |
| 2 |
10% NaOCl, DCM, rt, 1 h |
68 |
| 3 |
10% NaOCl, TEA, DCM, rt, 3 h |
52 |
The optimized conditions (aq. NaOCl in DCM at rt) for
5a synthesis were also applied to the synthesis of 7-(4-fluorophenyl)-, 7-(4-bromophenyl)- and 7-methyl-3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazoles
5b–
d to evaluate the scope of the methodology. The products were obtained in yields of 63%, 64% and 42%, respectively. In addition, we investigated whether the obtained 3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole system can be further oxidized. Several oxidation reaction conditions were tested, e.g.,
5a was stirred in DMSO at 110 °C in an open atmosphere [
56] or treated with a catalytic amount of Pd/C in acetic acid [
57]; the best result was obtained using activated MnO
2 as an oxidant in toluene in a Dean–Stark apparatus for 4 h at reflux temperature [
58]. Furthermore, 4
H,7
H-Pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole derivative
6 was formed in 38% yield.
A similar brief study on 5-chloropyrazole-4-carbaldehydes as synthons for intramolecular 1,3-dipolar cycloaddition was also reported by L‘abbé et al. [
59]. The authors noticed that 5-allyloxypyrazole-4-carbaldehyde derived from 5-chloropyrazole-4-carbaldehyde and further used as a precursor for intramolecular 1,3-dipolar cycloaddition reactions underwent a slow Claisen rearrangement to 4-allyl-5-hydroxypyrazole, even at room temperature. In contrast, we found 3-allyloxypyrazole-4-carbaldehydes to be stable. They can be stored in the laboratory at room temperature.
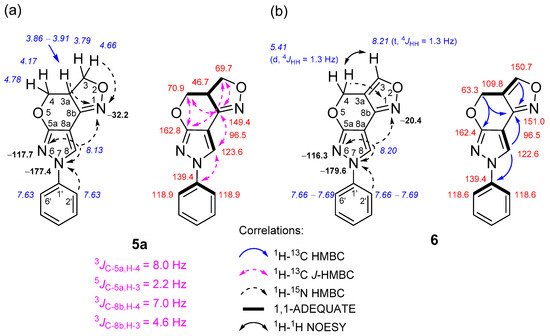
The formation of 3a,4-dihydro-3H,7H- and 4H,7H-pyrazolo[4′,3′:5,6]pyrano[4,3-c][1,2]oxazole ring systems was easily deduced after an in-depth analysis of NMR spectral data, which were obtained through a combination of standard and advanced NMR spectroscopy techniques, such as 1H-13C HMBC, 1H-13C J-HMBC, 1H-15N HMBC, 1H-13C HSQC, 1H-13C H2BC, 1H-1H COSY, 1H-1H NOESY and 1,1-ADEQUATE experiments (Figure 1).
Figure 1. Relevant 1H-13C HMBC, 1H-13C J-HMBC, 1H-15N HMBC, 1H-1H NOESY and 1,1-ADEQUATE correlations and 1H NMR (italics), 13C NMR and 15N NMR (bold) chemical shifts of compounds 5a (a) and 6 (b).
In the case of compound 5a, the multiplicity-edited 1H-13C HSQC spectrum allowed us to identify the pairs of geminally coupled methylene protons, since both protons displayed cross-peaks with the same carbon. For instance, it showed two pairs of negative signals at δH 4.66, 3.79 and 4.78, 4.17 ppm, which have one-bond connectivities with the methylene carbons C-3 (δ 69.7 ppm) and C-4 (δ 70.9 ppm), respectively. The chemical shifts of these methylene groups are expected to be similar and downfield compared to a neighboring methine group at site 3a, because both are bound to the oxygen atoms O-2 and O-5. This adjacent protonated carbon C-3a (δ 46.7 ppm) relative to the aforementioned methylene sites was easily assigned from an appropriate correlation in the 1H-13C H2BC spectrum.
In the 1H-15N HMBC spectrum of 5a, strong long-range correlations between the methylene 3-H proton at δ 4.66 ppm and the 3a-H proton at δ 3.86–3.91 ppm with the oxazole N-1 nitrogen at δ −32.2 ppm were observed. The lack of long-range correlations with another pair of methylene protons (δ 4.78, 4.17 ppm), and the aforementioned N-1 nitrogen, strongly hinted at assigning this methylene group to site 4. In order to unambiguously discriminate between these methylene groups, the 1H-13C heteronuclear couplings were measured using a 1H-13C J-HMBC experiment, thus providing complimentary evidence for correct structural assignment. The J-HMBC spectrum showed a strong correlation between the methylene proton δ 4.78 ppm and the quaternary carbon C-5a with an 8.0-hertz coupling constant, while the proton δ 4.66 ppm correlated very weakly, with a J value of only 2.2 Hz, which was attributed to a 5JC-5a, H-3. Finally, the pyrazole 8-H proton (δ 8.13 ppm) not only exhibited long-range HMBC correlations with neighboring N-7 “pyrrole-like” (δ −177.4 ppm) and N-6 “pyridine-like” (δ −117.7 ppm) nitrogen atoms, but also with the C-5a, C-8a and C-8b quaternary carbons, which were unambiguously assigned with the subsequent 1,1-ADEQUATE experiment, thus allowing all the heterocyclic moieties to be connected together. The structure of compounds 5b–d was determined by analogous NMR spectroscopy experiments, as described above. The skeleton of the pyrazolo[4′,3′:5,6]pyrano[4,3-c][1,2]oxazole ring system contains three nitrogen atoms. The chemical shifts of the N-1, N-6 and N-7 atoms of compounds 5a–c were in a range from δ −30.9 to −32.2, δ −116.9 to −117.7 and δ −177.4 to −179.5 ppm, respectively, while in the case of compound 5d, which lacked a phenyl moiety at site 7, the chemical shifts of N-1, N-6 and N-7 atoms were δ −35.8, δ −112.3 and δ −194.4 ppm, respectively.
In the case of compound
6, a comparison of the
1H NMR spectra between
5a and
6 clearly indicated the disappearance of methine 3a-H (δ 3.86–3.91 ppm) and methylene 3-H protons (δ 4.66 and 3.79 ppm) and the formation of a new downfield methine 3-H proton signal at δ 8.21 ppm. The aforementioned methine proton that appeared as a triplet was mutually coupled with methylene 4-H protons (doublet, δ 5.41 ppm), as indicated by their
meta-coupling (
4JHH = 1.3 Hz). Moreover, a comparison between the
1H-
1H COSY and
1H-
1H NOESY spectra showed a complete absence of COSY cross-peaks between 3-H and 4-H and only strong NOEs, which confirmed their proximity in space. This finding strongly hinted at a neighboring quaternary carbon at site 3a, which was unambiguously assigned from 1,1-ADEQUATE spectral data, where the protonated carbons C-3 (δ 150.7 ppm) and C-4 (δ 63.3 ppm) showed a sole correlation with C-3a at δ 109.8 ppm. As expected, the
15N chemical shifts of N-6 (δ −116.3) and N-7 (δ −179.6) atoms were highly comparable to those of compounds
5a–
c; only the N-1 atoms were slightly different and resonated at δ −20.4 ppm, which is in good agreement with the data reported in the literature [
60].
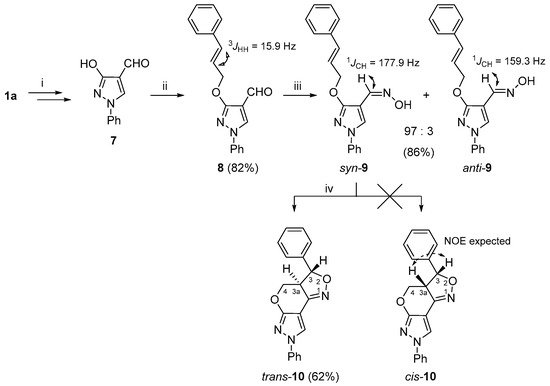
To expand the structural diversity of the obtained 3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole system, we prepared additional
vic-cinnamyloxy-oxime
9 as a substrate for the INOC reaction (
Scheme 2). As the cinnamyloxy group turned out to be sensitive towards Vilsmeier–Haack reaction conditions, the
O-alkylation formylation sequence of compound
1a successfully applied to the synthesis of 3-allyloxypyrazole-4-carbaldehydes
3a–
d was reorganized. In short, first, the hydroxy group of pyrazol-3-ol (
1a) was transformed to a benzyloxy group; then, the obtained 3-benzyloxypyrazole was formylated under the Vilsmeier–Haack reaction conditions, and the protecting OBn group was cleaved by TFA to give 3-hydroxy-1
H-pyrazole-4-carbaldehyde
7 [
35]. The latter compound was subjected to an alkylation reaction with cinnamyl chloride and the appropriate 3-cinnamyloxy-1
H-pyrazole-4-carbaldehyde (
8) was obtained in very good (82%) yield. A subsequent reaction of
8 with hydroxylamine gave the aldoxime
9, which was successfully used for the INOC reaction, and 3-phenyl-3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole
trans-
10 was obtained with a fair (62%) yield.
Scheme 2. Synthetic route for the 3a,4-dihydro-3
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole
10. Reagents and conditions: (i) in accordance to ref. [
35]; (ii) NaH, DMF, 0 °C, 15 min, cinnamyl chloride, 60 °C, 15 min; (iii) NH
2OH·HCl, NaOAc, EtOH, reflux, 15 min; (iv) NaOCl, DCM, rt, 1 h.
While the structural elucidation of compound
trans-
10 was straightforward and followed the same logical approach as in the case of compounds
5a–
d and
6, determination of the relative configuration at C-3 and C-3a proved to be a more challenging task and was achieved by combined analysis of NOESY,
J-coupling and molecular modeling data. For instance, the initial geometry optimizations were performed using MM2 and MMFF94 force fields [
61], followed by DFT methods using B3LYP/def2-TZVP, as implemented in ORCA 5.0.0 [
62], which provided the dihedral angle values between H-C(3)-C(3a)-H for structures
trans-
10 (154.34°) and
cis-
10 (19.28°). Then, the theoretical
1H-
1H coupling constants were calculated with the same software package following a standard procedure using a B3LYP/PCSSEG-2 basis set. The dihedral angle values were used in the calculation of
3JH3,H3a by the Haasnoot–de Leeuw–Altona (HLA) equation [
63]. The
3JH3,H3a values estimated by the HLA method were 10.0 Hz for
trans-
10 and 8.2 Hz for
cis-
10, while ORCA 5.0.0 calculations were 13.4 and 10.8 Hz, respectively. The experimental value 13.1 Hz, which was obtained from the
1H NMR spectrum, hinted in favor of the
trans-
10 structure. A highly similar class of heterocycles, naphthopyranoisoxazolines, were synthesized by Liaskopoulos et al. [
64], where the target compounds possessed a
trans configuration, as confirmed by X-ray and NMR analyses, and their
3JH3,H3a values were in the range of 12.2–12.5 Hz. Finally, unambiguous confirmation of
trans-
10 assignment was obtained from the
1H-
1H NOESY spectrum, as it was evident from the geometrically optimized structures (
Figures S80 and S81) that, in the case of
cis-
10, there should be a strong NOE between protons 3-H and 3a-H, while the NOE between 3a-H and the neighboring 3-phenyl group aromatic protons is not possible. However, in our case, the
1H-
1H NOESY spectrum showed completely opposite measurements. Moreover, a distinct NOE between protons 3-H/4-H
a and 3a-H/4-H
b is only possible if the relative configuration is
trans-
10.
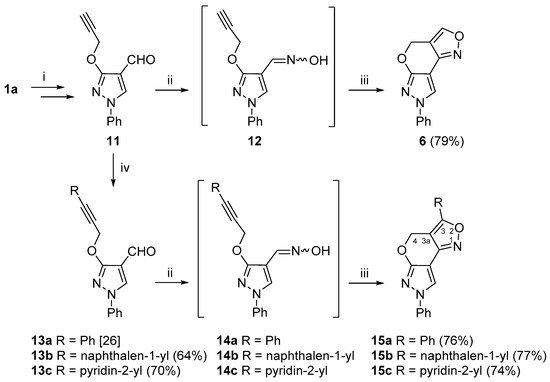
We also investigated the INOC reaction of
vic–alkyne–oxime substrates
12 and
14a–
c (
Scheme 3). To obtain the intermediate compound
12, firstly, 3-hydroxypyrazole
1a was
O-propargylated and formylated to give carbaldehyde
11 [
26]. Compound
11 was then successfully converted to 4
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole
6 via the INOC reaction of intermediate oxime
12, and the targeted new polyheterocyclic compound
6 was obtained in good (79%) yield. In addition, alkyne
11 was further subjected to the Sonogashira cross-coupling reaction with various (het)arylhalides, i.e., iodobenzene, 1-iodonaphthalene and 2-bromopyridine, under the standard Sonogashira cross-coupling reaction conditions (Pd(PPh
3)
2Cl
2, CuI, DMF, 60 °C, argon atmosphere) to give alkynes
13a–
c in good yields [
26]. Compounds
13a–
c were further treated with hydroxylamine hydrochloride to provide aldoximes
14a–
c, which were used in the INOC reaction without further purification. Aldoxime
14a was subjected to a detailed NMR analysis, and, to our delight, it was obtained as a pure
syn isomer, which was easily elucidated from a
1JCH coupling constant of the iminyl moiety, which was 179.2 Hz. Moreover, 4
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazoles
15a–
c were obtained in good yields.
Scheme 3. Synthetic route for the 4
H,7
H-pyrazolo[4′,3′:5,6]pyrano[4,3-
c][1,2]oxazole ring system. Reagents and conditions: (i) in accordance to ref. [
26]; (ii) NH
2OH·HCl, NaOAc, EtOH, reflux, 15 min; (iii) NaOCl, DCM, rt, 1 h; (iv) RX, Pd(PPh
3)
2Cl
2, TEA, DMF, 60 °C, 15 min.
As expected, the chemical shifts of the 3-aryl-substituted compounds 15a–c were highly similar to those of compound 6. A distinct difference in the 1H NMR spectra of the aforementioned compounds was that they contained only a singlet for the methylene 4-H protons in the area of δ 5.32–6.03 ppm, which indicated the lack of coupling partners. The data from the 1H-13C HMBC spectra revealed a distinct long-range correlation between the aforementioned methylene protons and a quaternary carbon at site 3. Moreover, the protons from a neighboring 3-aryl moiety shared an HMBC cross-peak with carbon C-3 as well, thus allowing different structural fragments to be joined together. The chemical shifts of the N-1, N-6 and N-7 atoms of 3-aryl-substituted compounds were in ranges of δ −23.9 to −25.0, δ −116.4 to −117.4 and δ −179.6 to −180.1 ppm, respectively, while, in the case of compound 15c with a pyridin-2-yl moiety, the pyridine nitrogen resonated at δ −72.8 ppm.
3. Conclusions
This study has developed a convenient method for the preparation of 3a,4-dihydro-3H,7H- and 4H,7H-pyrazolo[4′,3′:5,6]pyrano[4,3-c][1,2]oxazoles from easily obtainable 3-(prop-2-en-1-yloxy)- or 3-(prop-2-yn-1-yloxy)-1H-pyrazole-4-carbaldehydes by INOC reaction of intermediate oximes. The key stage—nitrile oxide preparation from the corresponding aldoximes—was carried out by oxidation with sodium hypochlorite. The method was applied for the synthesis of pyrazolo[4′,3′:5,6]pyrano[4,3-c][1,2]oxazoles with various substituents in the third or seventh position. In addition, extensive NMR spectroscopic studies have been undertaken using standard and advanced methods to unambiguously determine the configuration of intermediate aldoximes, showing the predomination of the syn-isomer, as well as the structure of new polycyclic systems.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26185604