Heart failure (HF) studies are mostly designed to prove the efficacy of therapeutic interventions in patients with a predefined cut-off value of left ventricular ejection fraction (LVEF) as primary entry criteria. On the other hand, a predefined low value of the glomerular filtration rate (GFR) may be applied as an exclusion criterion. Thus, on one side, this decision-making process recognizes HF as a “cardiorenal condition”, while on the other side, investigation of HF therapies’ aim to establish their benefit on cardiac outcome, while renal outcomes are often not rated as a concurrent target.
Recently, the gliflozins, inhibitors of sodium-glucose co-transporter 2 (SGLT2i), were cleaved in the cardiovascular arena, proving unexpected effectiveness in restraining HF exacerbations, while they were investigated as hypoglycemic drugs in type 2 diabetes mellitus patients (T2DM)
[1]. The pharmacologic action of this class of drug is to inhibit the reabsorption of Na
+ and glucose in the proximal segment of the glomerular tubule; therefore, they work with exquisite renal effect. In the following studies, the extension of benefit of those molecules on HF outcomes was coupled with concurrent decrease in renal adverse outcomes in a fashion that made clear the need to overcome the heterogeneity in the reporting of kidney function, kidney outcomes, and definitions for kidney end-points in clinical trials. This step is now mandatory in order to gauge the benefit of interventions across large-scale studies performed in T2DM, chronic kidney disease (CKD) and HF
[2].
2. Body Fluid Homeostasis Is Centered on Kidney Function
The renal fluid and electrolytes excretion allow the adaptation of body fluid and electrolytes content to the physiological needs. The body fluid balance is intrinsically connected with Na+ Cl− K+ homeostasis that is, in turn, locally involved in preserving kidney vitality and function.
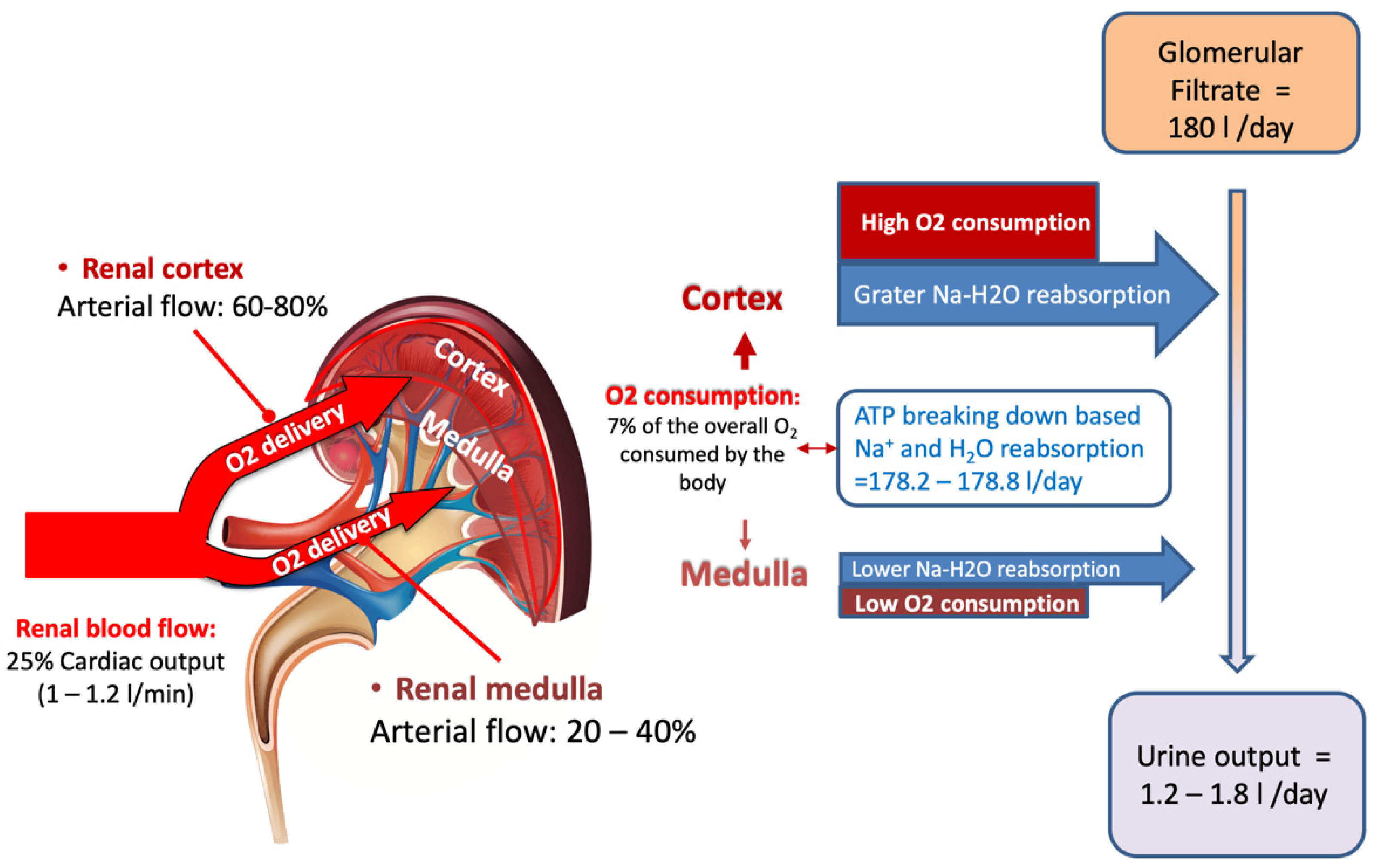
The kidney represents only 0.5% of total body weight receiving, albeit with ~25% of cardiac output
[3], while its physiologic function needs to extract only 10–15% of available oxygen supply (corresponding to the 7% of the whole O
2 body consumption)
[4]. Thus, a huge disparity clearly exists between renal mass, renal O
2 consumption and, more so, the amount of delivered renal O
2.
Indeed, only a minor portion of the very high amount of arterial blood delivered to the kidney contributes to the glomerular filtrate generation. In an animal model, the renal fractional extraction of O
2 declines when renal blood flow increases, while renal parenchymal O
2 remains unchanged
[5], unveiling the function of the unique arrangement of the preglomerular vessels architecture. This specific arrangement presides over the diffusional shunting from arteries to veins, restricting the blood flow directed toward the glomerular vasculature. This mechanism contributes to the dynamic regulation of intrarenal oxygenation and directly affects the GFR generation with proportional increase in the filtered Na
+.
A further step in regulation of the GFR generation takes place in the glomerular vasculature that provides hydraulic regulation of pressure in and out of the glomerulus, and that is prominently based on neural endocrine response
[6].
Due to the complex renal hemodynamic regime, an intrarenal metabolic control with specific effects on the vasculature and transport systems of cortex and medulla is at the base of the kidney function
[4][6]. Many small molecules with short action duration, generated by local demand (nitric oxide, bradykinin, endothelin, angiotensin II, and prostacyclin), concur with local signaling cascades regulating renal function
[6].
In the kidney, the largest amount of O
2 consumption occurs in the cortex, where the blood flow is prominently directed to generate the GFR that, in turn, is linked to the Na
+ reuptake in the proximal tubular segment. The reabsorption process is based on ATP utilization. In the normal subject the average GFR is 180 L/day while the normal urine output ranges from 1.2 to 1.8 L/day, meaning that roughly 1% of the glomerular filtered load is excreted
[7] (
Figure 1).
Figure 1. Despite renal mass accounts only for 0.5% of total body mass, 25% of cardiac output is delivered to the kidney, which consumes the 7% of the whole body oxygen consumption. Renal oxygen consumption is mostly connected to the selective reabsorption of glomerular filtrate that corresponds to approximately to 180 L/day, while daily urine output is restricted to 1.2–1.8 L/day. The largest reabsorption work takes place in the proximal segment of glomerular tubule in the cortex area, where arterial blood flow provides the highest oxygen delivery to supply energy for urine concentration and selective solutes excretion (see text for details).
Normal daily glomerular filtrate contains ~1 mol (~180 g) of glucose
[3] that, if eliminated into the urine, would generate an energy cost equivalent to ~1/3 of the body’s total caloric expenditure. Conversely, in the S1 segment of the glomerular proximal tubule the type 2 sodium-glucose co-transporter (SGLT) 2 reabsorbs 80–90% of the filtered glucose, coupled one to one with Na
+. The remaining 10–20% of glucose is reabsorbed by SGLT1 in the S2/S3 segment by coupling one sugar molecule with two Na
+ atoms. Thereby, SGLT1 uses as much as twice the energy per one reabsorbed glucose molecule as SGLT2. The synergy of both sodium-glucose co-transporters accounts for a maximum reabsorption capacity of ~2.5 mol (450 g) of filtered glucose per day
[7].
As SGLT1’s reuptake of glucose requires double the Na
+ as SGLT2, the higher maximum tubular transport of glucose not only entails higher exposure to hyperglycemia but also disposes to fluid retention
[8].
The activation of all those sophisticated mechanisms linked to sodium and glucose reabsorption highlights the amount of energy consumed by the selective regulation of solutes excretion and by the reabsorption of Na+ and water from the glomerular filtrate.
This energy consuming process makes the GFR production the major determinant of kidney O
2 consumption, strongly connected to the entire renal metabolism
[6][8][9].
This high-energy process leaves less oxygen available to the medullar vasculature
[3][6]. As a consequence, the GFR, with the connected tubular Na
+ load, affects the O
2 available for transport work in the cortex and, more critically, in the medulla.
These processes must be tightly coordinated to avoid fluid and electrolyte losses and the intrarenal mechanisms driving the punctual adjustment of the single glomerulus function, include glomerulotubular balance and tubule-glomerular feedback (TGF)
[6].
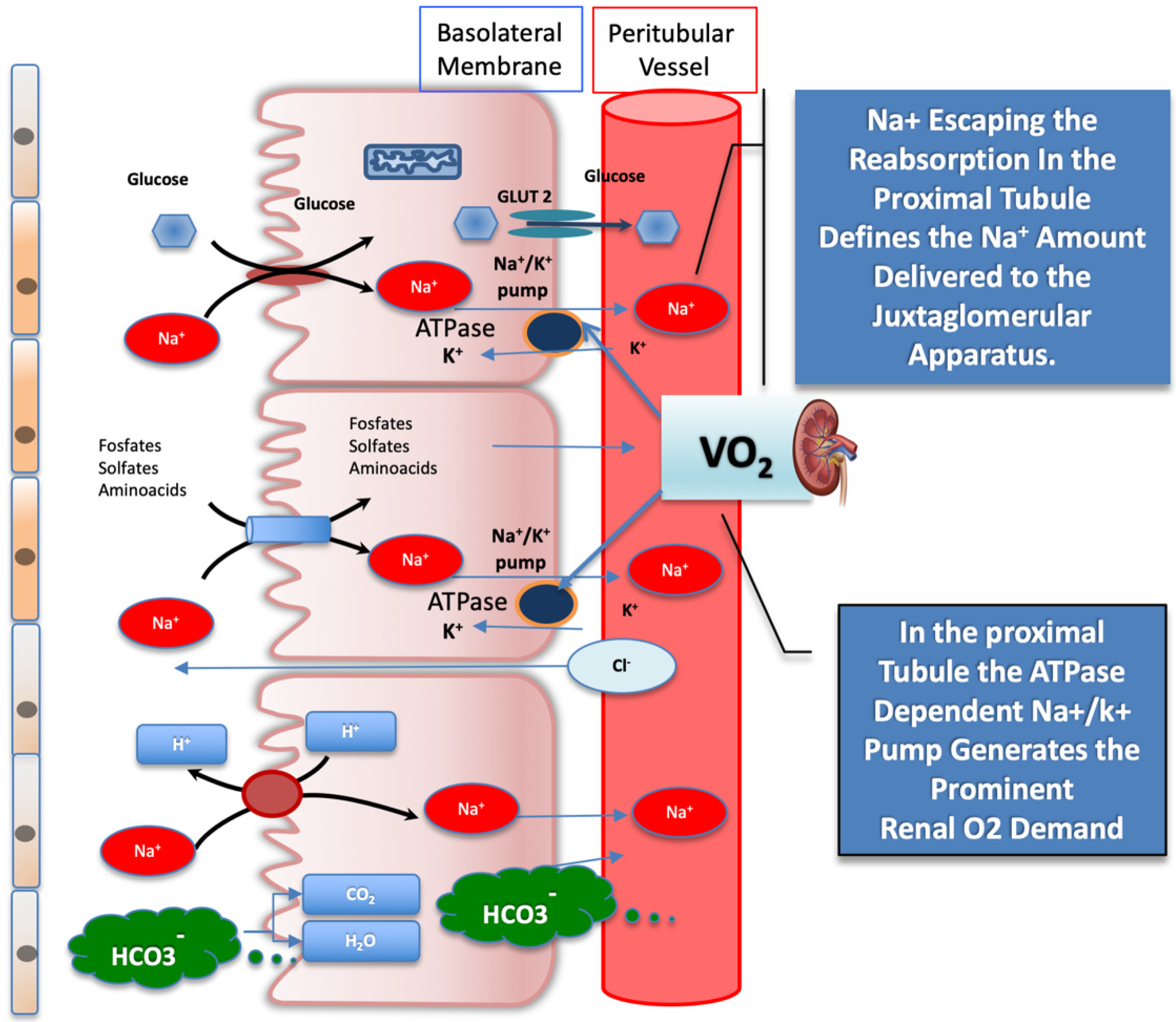
Any increase in the reabsorption process, accounting for the glomerulotubular balance mechanism, is linked to the plasma oncotic pressure resulting after filtration, driven by the intraglomerular pressure gradient. The glomerulotubular balance mechanism takes place almost entirely in the proximal tubule and in the cortical thick ascending limb of Henle, locally involving the highest O
2 demand driven by the sodium–potassium ATPase, which provides sodium potential to reabsorb solutes from the filtrate
[4][6] (
Figure 2).
Figure 2. Schematic illustration of filtrates reabsorbed from the glomerular proximal tubule activated by sodium-glucose co-transporter (SGLT) 2, which operates in the glomerular proximal tubule. The SGLT2, the bicarbonate reabsorption, and the Na+/K+ ATPase, which provides sodium energy to drive both, are represented. The active reuptake of bicarbonate and of other solutes, coupled with Na+, drives the highest tissue O2 consumption in the kidney. The amount of Na+ escaping the reabsorption in the proximal section tubule is sensed by the juxtaglomerular apparatus and sets the afferent arteriole tone. See text for details. GLUT2 = glucose transporter 2; SGLT2 = sodium-glucose co-transporter 2.
It is important to note that the SGLT1 protein is also present in the small intestine, where it acts as rate-limiting factor for absorption of glucose and galactose, by using the transmembrane sodium gradients to drive the cellular uptake of these molecules. In the multiethnic population of the Atherosclerosis Risk in Communities (ARIC) study, it was observed that a specific modification of the SGLT1 haplotype, qualified by the sequence N51S/A411T/H615Q, was associated with protection from postprandial hyper-glycemia. Those data support the hypothesis the SGLT1 inhibition may result in the decline of T2DM, HF, and mortality incidence by restraining the postprandial glucose levels in subjects at risk
[10].
3. Sodium-Glucose Co-Transporters Over-Expression: The Related Consequences
In individuals with diabetes mellitus, as Na
+ and glucose are co-transported, the enhanced reabsorption of glucose is coupled with higher body sodium content, and not surprisingly, 60 to 70% of patients with T2DM may become hypertensive
[11]. Consistently in persons with diabetes, the increased exposure of proximal tubular cells to filtered glucose promotes SGLT2 overexpression with further enhancement of glucose reabsorption, leading to a paradoxical increase in the urinary glucose excretion threshold
[7][12]. Moreover, the persistence of an uncontrolled glycemic ambient induces the increase in SGLT1/2 mRNA expression by 36% and 20%, respectively, leading to further Na
+ load reabsorption. In this circumstance, SGLT1/2 activity may account for as much as more than 14% of renal Na
+ reabsorption along the whole tubule
[12], with a significant dip in Na
+ load reaching the JGA. The decreased amount of Na
+ passing through the JGA not only reduces local adenosine production, but also attenuates the control on renin angiotensin II axis. In turn, AT II not only restrains the efferent arteriole section, but also reduces the glomerular capillary surface area, impacting hydraulic glomerular conductivity with convergent mechanisms in increasing glomerular filtration pressure
[13][14] (
Figure 3).
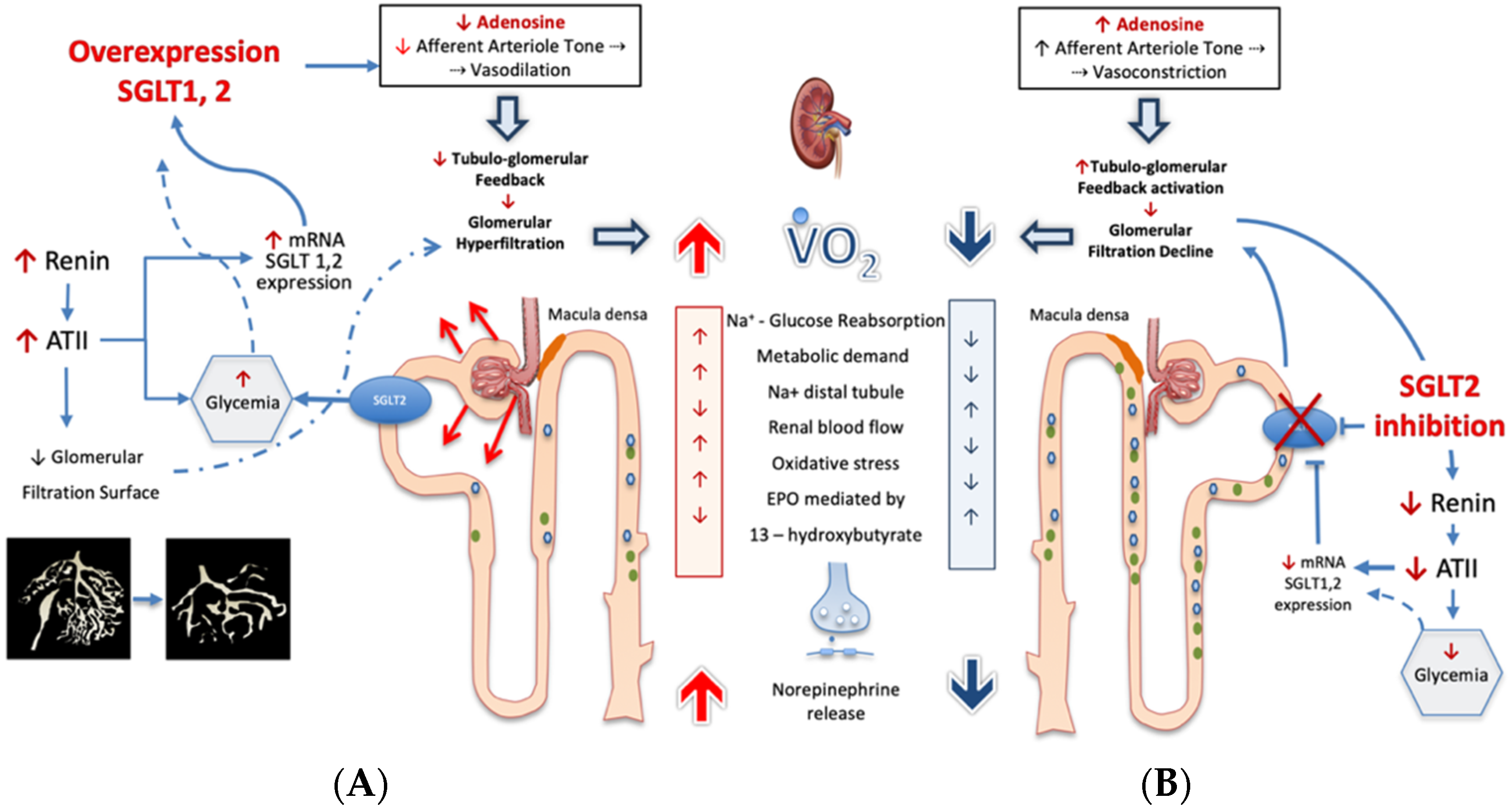
Figure 3. Under conditions of ambient hyperglycemia (panel (
A)), sodium-glucose co-transporter 2 activity (SGLT2) is increased, thereby reducing the juxtaglomerular apparatus delivery of Na
+. This affects the tubuloglomerular feedback mechanisms, as the concentration of Na
+ that transits across the juxtaglomerular apparatus establishes the adenosine triphosphate (ATP) release and its breakdown to adenosine. Local adenosine concentration acts by vaso-constricting afferent arteriole. Thus, any adenosine concentration decrease may impact the appropriate regulation of glomerular blood flow and of glomerular filtration pressure and, therefore, of renal metabolic work. An unfavorable consequence of the higher metabolic work is the increased cortical oxidative stress-induced tubulointerstitial damage that causes a fall in erythropoietin production
[9]. The adenosine generation in the juxtaglomerular apparatus is linked to a large network of biological effects such as the control of renin release; therefore, a decreased breakdown of ATP in the juxtaglomerular apparatus may lead to enhanced ATII generation. Angiotensin II not only affects the circulation balance systemically, but also compromises the glomerulotubular balance by restraining the efferent arteriole section and the glomerular capillary surface area. All together, these responses account for the incremental neural transmitter release, leading to the exceedingly high renal norepinephrine spillover occurring in heart failure progression
[15]. Under these conditions, the SGLT2 inhibition (
B), by inducing glucoresis and natriuresis, is able to re-establish the distal tubular flow and Na
+ delivery to the juxtaglomerular apparatus. In this way, SGLT2 inhibition restores local adenosine generation that reestablishes the afferent arteriole tone, leading to lower filtration pressure and to lower metabolic work while it restores control on neuroendocrine pathway. See text for details. SGLT2 = sodium-glucose co-transporter-2; VO2 = Oxygen consumption; ATII = angiotensin II; EPO = erythropoietin.
In general, the accepted marker of high intraglomerular pressure is the glomerular hyperfiltration defined as a GFR ≥ 135 mL/min·1.73 m
2: beyond this value, glomerular hyperfiltration hurts the mesangial structure, allowing albumin leakage in the filtrate and paving the way toward end-stage diabetic nephropathy
[12][13][14].
In addition to these adverse actions, angiotensin II enhances glycemia together with SGLT2 expression in proximal tubular cells, contributing to the metabolic derangement
[14]. In the long run, the intraglomerular hemodynamic changes cause profound glomerular damage, leading to progressive CKD and deeply impairing the systemic hemodynamic balance. Indeed, the higher Na
+ proximal tubule reabsorption draws further water, adding to passive sodium reabsorption that aggravates fluid retention and tissue congestion
[8]. The mechanisms concur to explain why local kidney regulatory action may become a critical aspect of renal perfusion in the HF syndrome independently by coexistent diabetes. Low cardiac output results in arterial vasculature under-filling, causing arterial blood to be shunted from the kidneys to the systemic circulation. The disproportionate decrease in renal fraction, secondary to cardiac output, critically enhances the renal efferent sympathetic activity
[16]. In this condition, norepinephrine spillover strikingly increases in the kidney, becoming a prominent index of unfavorable outcome independent of left ventricular performance, GFR and overall sympathetic activation
[15]. Norepinephrine has a potent action in enhancing peripheral vascular resistances restricting end-organs perfusion while it enhances the glycemic level, leading to increased SGLT2 expression in the nephron and insulin secretion. In two cohort studies performed in patients hospitalized for acute decompensated HF, the blood glucose levels were linked to worse outcomes, irrespective of the diabetes status. In the first study, at a median follow-up of 1.8 years, a greater risk of mortality was present in subjects with higher glucose level at hospital admission in comparison to patients without either blood glucose elevation or diabetes
[17]. In a second cohort study, the occurring glycemic variability, but not mean hospital glucose level, was associated with inpatient mortality
[18]. Pathophysiology and investigational data link neurohormonal changes and failing glycemic control to HF progression, whether or not diabetes is diagnosed.