Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Thermosensitive liposomes (TSL) belong to the category of triggered nanoparticle drug delivery systems (DDS) where a drug associated with the DDS is released in response to an external trigger. TSL are triggered by heat and release the encapsulated drug when exposed to mild hyperthermia (HT), typically ~40–43 °C. TSL were first described more than four decades ago.
- thermosensitive liposomes
- hyperthermia
- cancer
- nanoparticles
- drug delivery systems
1. Introduction
Thermosensitive liposomes (TSL) belong to the category of triggered nanoparticle drug delivery systems (DDS) where a drug associated with the DDS is released in response to an external trigger [1][2][3][4]. TSL are triggered by heat and release the encapsulated drug when exposed to mild hyperthermia (HT), typically ~40–43 °C. TSL were first described more than four decades ago [5][6][7][8]. Since then, numerous TSL formulations combined with various drugs have been described, as summarized in prior reviews [9][10][11][12][13][14][15]. TSL are most often administered systemically, e.g., by intravenous infusion, and then circulate in the blood stream for an extended duration. Combined with localized hyperthermia, TSL enable loco-regional drug delivery (Figure 1). This enables the delivery of a large drug dose to a targeted tissue region (e.g., tumor) while reducing systemic toxicities. Therefore, TSL are attractive as a therapeutic strategy in cancer patients where loco-regional drug delivery is beneficial, but less useful in metastatic cancer patients that require systemic therapy. While TSL have been most widely investigated for drug delivery in cancer therapy, additional potential clinical applications include the delivery of antibiotics [16][17], the treatment of inflammatory diseases [18], and the treatment of blood clots [19].
TSL enable two different delivery approaches: extravascular triggered release, and intravascular triggered release (Figure 2) [14][20][21][22][23]. Extravascular triggered release requires the extravasation of the TSL, followed by HT-triggered release of the encapsulated agent [14][24]. This extravasation is based on TSL accumulation within the tumor interstitium facilitated by enhanced permeability and retention (EPR) [14][25][26][27][28]. Several papers described the limitations of the EPR effect, such as high intra- and inter-tumor variability, and an apparent upper delivery limit [14][27][29]. Some reviews highlight the need for delivery strategies that do not rely on EPR [14][27][29].
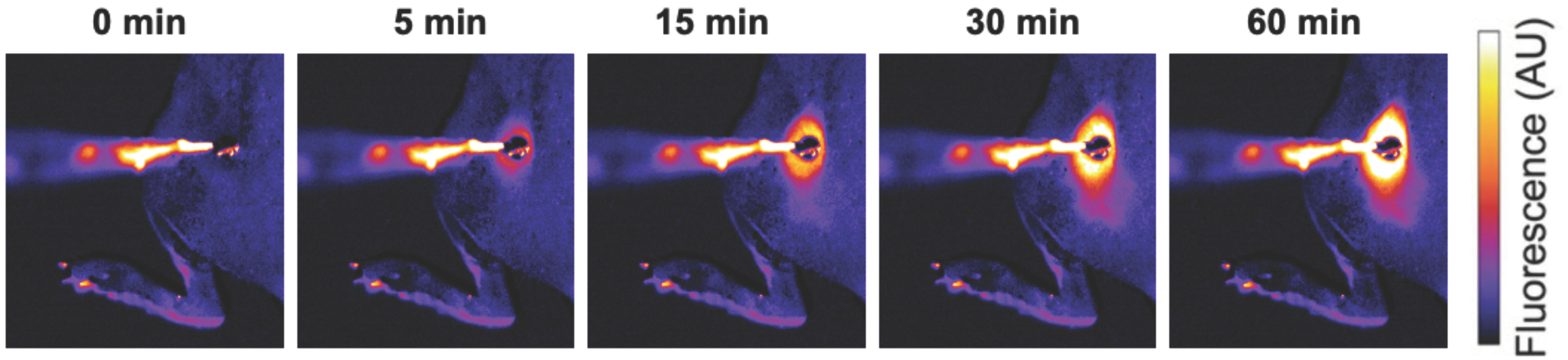
Figure 1. Localized drug delivery with thermosensitive liposomes (TSL). Following the administration of TSL-encapsulated doxorubicin (Dox), a subcutaneous mouse tumor was heated by a surface heating probe to 43 °C. Fluorescence imaging during hyperthermia visualizes the localized delivery of the fluorescent drug (Dox). Drug delivery takes place as long as hyperthermia is applied, here visualized by a fluorescence increase over the 60 min heating duration. Figure reproduced from [30] (published under Creative Commons CC BY license).
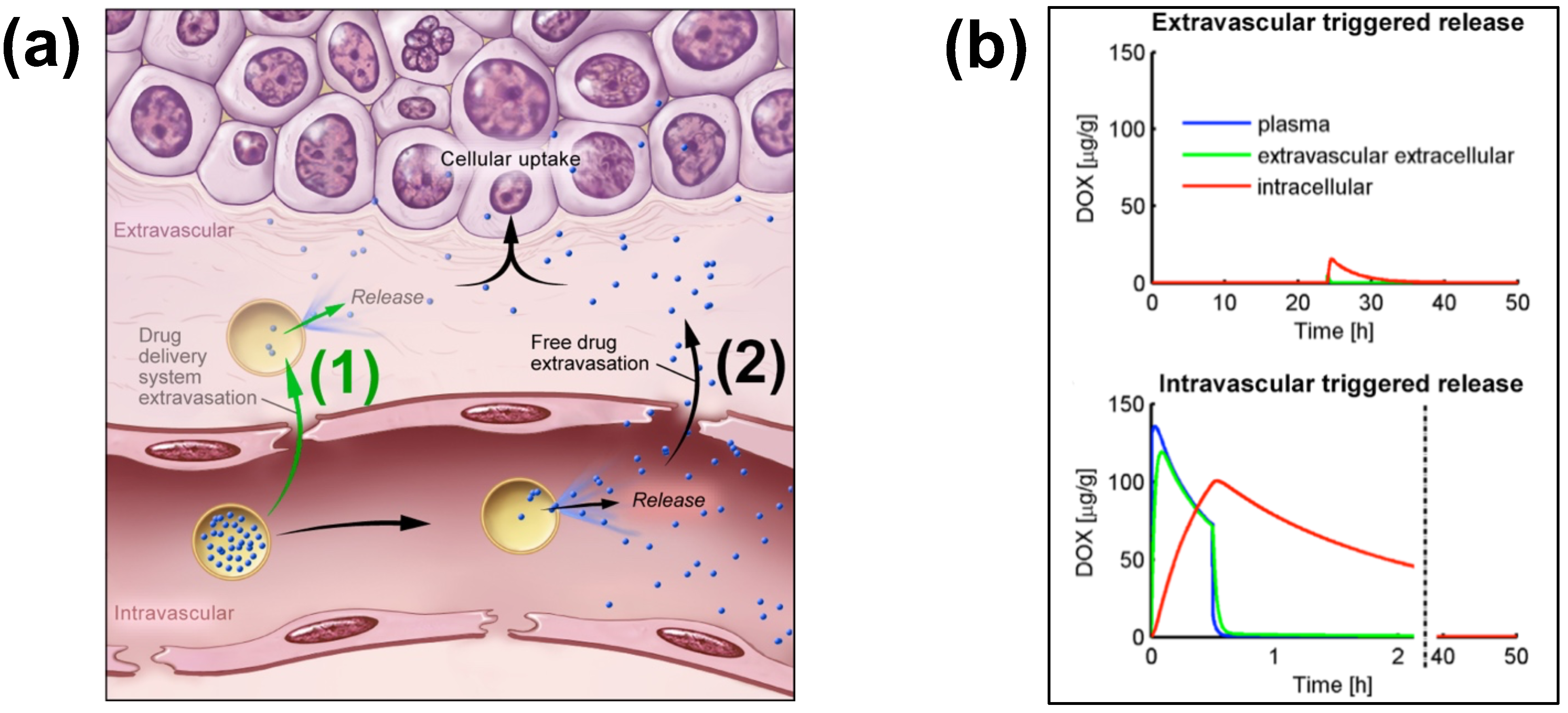
Figure 2. Extra- and Intra-vascular triggered release. (a) (1) Traditionally, nanoparticle DDS have been based on passive tumor targeting due to enhanced permeability and retention (EPR), where drug is released following extravasation of the DDS. (2) For TSL with intravascular triggered release, EPR is not relevant: TSL enter the tumor microvasculature of the target region where the release trigger (i.e., hyperthermia) is present, and release the contained drug within the vasculature. The released drug extravasates rapidly into tissue and is then taken up by cancer cells. (b) Top graph: Concentration dynamics in plasma, interstitial, and intracellular compartments during extravascular triggered release. TSL were allowed to accumulate for 24 h in the tumor based on EPR, followed by hyperthermia triggered release. Bottom graph: Concentration dynamics during intravascular triggered release. Hyperthermia (30 min) was applied immediately after TSL administration. Concentration increases in plasma due to drug release. Released drug then extravasates into interstitium (extravascular extracellular space), where it is taken up by cells. Figure 3a reproduced from [20] (published under CC BY 4.0 license). Figure 3b reproduced from [22] (published under CC0 license).
Intravascular triggered release is a strategy where drug release occurs in the microvasculature while the TSL pass through the heated tumor, and does not require the EPR effect (Figure 2a) [6][20][21][22][23]. Many of the more recent TSL formulations are based on intravascular triggered release, and such TSL have demonstrated superior delivery efficacy, with up to 25× higher drug delivery compared to unencapsulated drugs [31]. Compared to non-triggered nanoparticle drug delivery systems, TSL based on intravascular triggered delivery demonstrate superior tumor drug uptake (Figure 3). In addition, the direct comparison of TSL with extra- versus intra-vascular triggered delivery strongly suggests that the latter is superior [22][24][32][33] (Figure 2b).
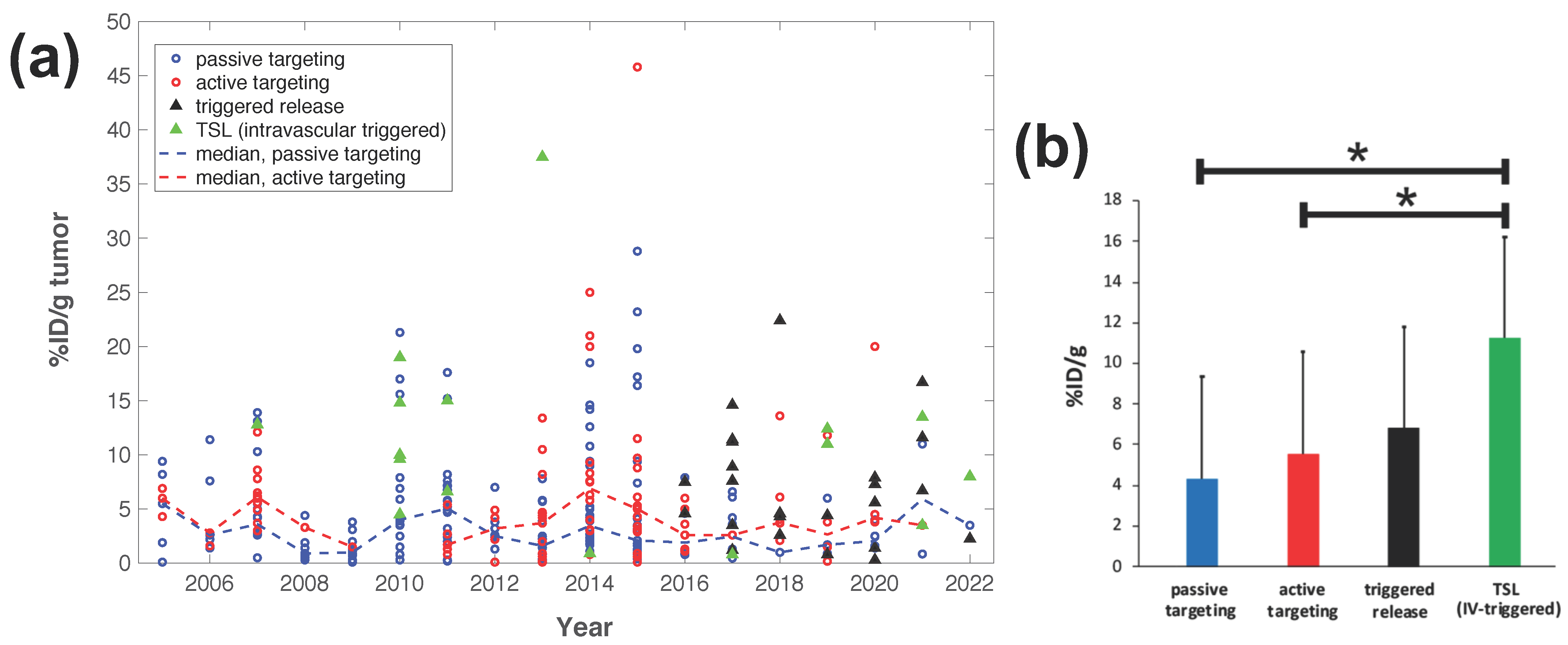
Figure 3. Delivery efficacy of intravascular triggered TSL compared to other nanoparticle DDS. A prior review compared the efficacy of 117 nanoparticle DDS studies published between 2005–2015 [27], and researchers combined data from this prior research to include studies published between 2016–2022 based on the same search algorithm [30][31][32][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114]. (a) Plot showing the delivery efficacy (%injected dose per gram tumor (%ID/g tumor)) based on the combined data [27][30][31][32][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114]. Each marker represents a published study, and dashed lines indicate the annual median for DDS with passive and active targeting. (b) The means of all prior studies in each category between 2005–2022 are compared, suggesting superior delivery efficacy of intravascular triggered TSL (* indicates statistical significance (p < 0.05)).
Tissue Transit Time
For TSL based on intravascular triggered release, the dynamics of blood flow through the tumor vasculature is of primary relevance. Blood/plasma with TSL enter a tumor segment through a supplying artery, pass through tumor capillaries, and exit the tumor segment through a draining vein. The average time that plasma spends within a tumor segment is termed the ‘tissue transit time’ (TT) (compared to plasma, red blood cells move significantly slower through capillaries, and thus remain for longer within the tumor segment [115]). The drug release from TSL, and drug extraction by tumor tissue, can only occur during this tissue transit time. Figure 4 visualizes the transit time between supplying artery and draining vein of a small mouse tumor segment. In human tumors, the mean transit time through a tumor has been measured for various tumor types. This mean tumor transit time varies widely, and is ~2 s for primary hepatocellular carcinoma [116], ~3 s for head and neck and prostate tumors [117][118], ~11 s for renal cell carcinoma [119], ~25 s for metastases to the liver [116], and ~30 s for breast cancer [120]. Furthermore, transit time and perfusion vary spatially within tumors such that transit time can be locally within a tumor considerably higher or lower than these mean values that were averaged over the whole tumor.
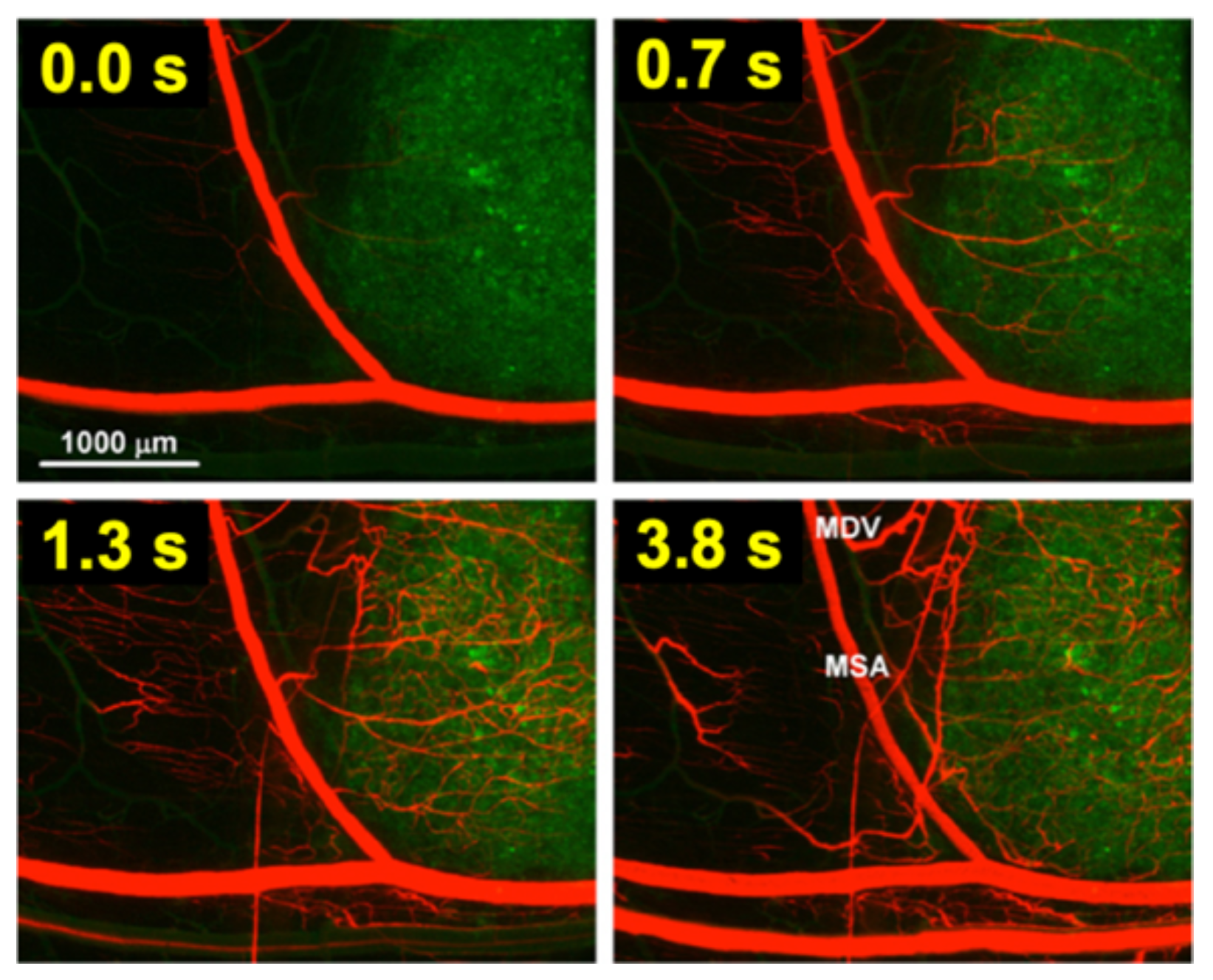
Figure 4. Tumor plasma transit time. Tumor (green fluorescent labeled cancer cells) was imaged by intravital fluorescence microscopy. A red fluorescent contrast agent was injected as bolus. The time at left upper corner of each image indicates timing relative to plasma first entering the tumor segment; plasma exits the tumor segment again within ~4 s (note: red blood cells move slower than plasma and remain longer in the tumor segment). In the final image (right lower corner), the main supplying artery (MSA), and main draining vein (MDV) of the imaged tumor segment are labeled. Figure reproduced with permission from [115].
As plasma with TSL enters capillary vessels within a heated tumor region, the TSL start releasing the drug and the drug is then extracted by the tumor (Figure 5b,c). Therefore, the plasma drug concentration varies along the vasculature as plasma flows between the supplying artery and draining vein of a tumor segment—in other words, a concentration gradient develops along the tumor microvasculature between the supplying artery and draining vein. Figure 5 illustrates schematically this microvascular concentration gradient along a representative capillary connecting the supplying artery and the draining vein of a tumor segment. For free (unencapsulated) drug, plasma drug concentration decreases as drug is extracted (Figure 5a). For TSL, drug is first released by hyperthermia, and then the released (free) drug is extracted (Figure 5b,c). Ideally, TSL completely release the encapsulated drug during the transit time to maximize tumor drug uptake—i.e., TSL that release their drug within seconds are preferable (Figure 5b).
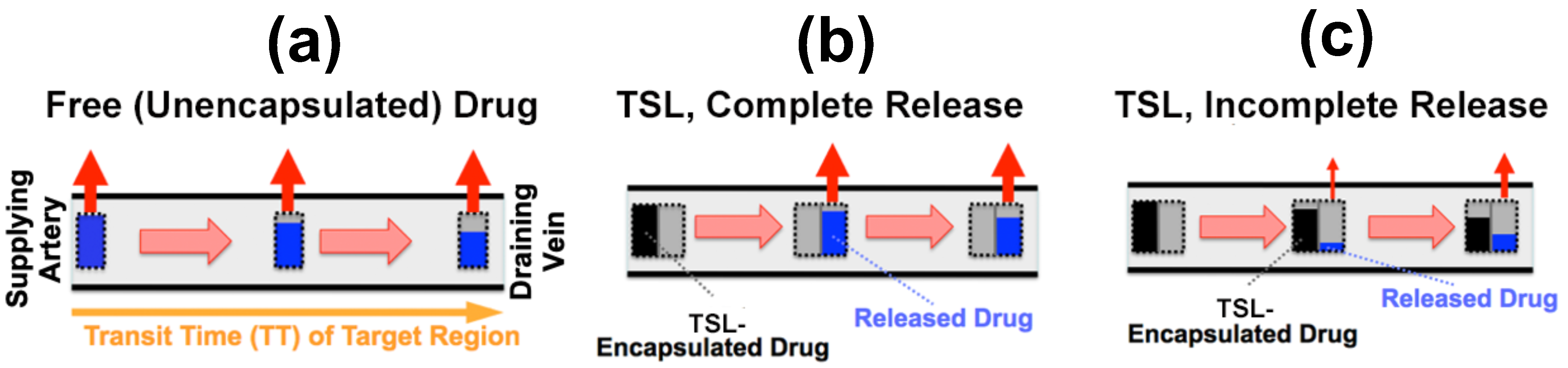
Figure 5. Microvascular concentration gradient. Plasma flows within a representative capillary between the supplying artery and draining vein of a tumor segment. Plasma concentration of unencapsulated/released drug (blue bar), and TSL-encapsulated drug (black bar) are shown, with red arrows indicating tumor drug uptake (i.e., drug extraction). Three cases are presented: (a) Unencapsulated drug infusion into supplying artery, (b) TSL with complete release during transit, and c) TSL with incomplete release during transit. In (b,c), drug is first released from TSL, followed by tissue uptake. Note that all figures show first pass where no drug is yet present in the tissue interstitium. Figure reproduced from [20] (published under CC BY 4.0 license).
2. Impact of TSL Properties on Drug Delivery
The methods for preparation and loading of various TSL formulations with different agents has been reviewed extensively in prior reviews [9][10][11][12][13][14][15]. Additionally, the factors and mechanisms that affect drug release from TSL have been summarized in detail in earlier publications [9][11][12]. Here, how TSL properties such as release kinetics and plasma stability affect drug delivery is focused on. These properties depend both on the TSL formulation and the drug. For example, the same formulation will have varying release kinetics depending on which drug is encapsulated [121]. In addition, the buffer used to measure release affects release kinetics [121], highlighting the importance of selecting an appropriate buffer (e.g., plasma) (Figure 6d).
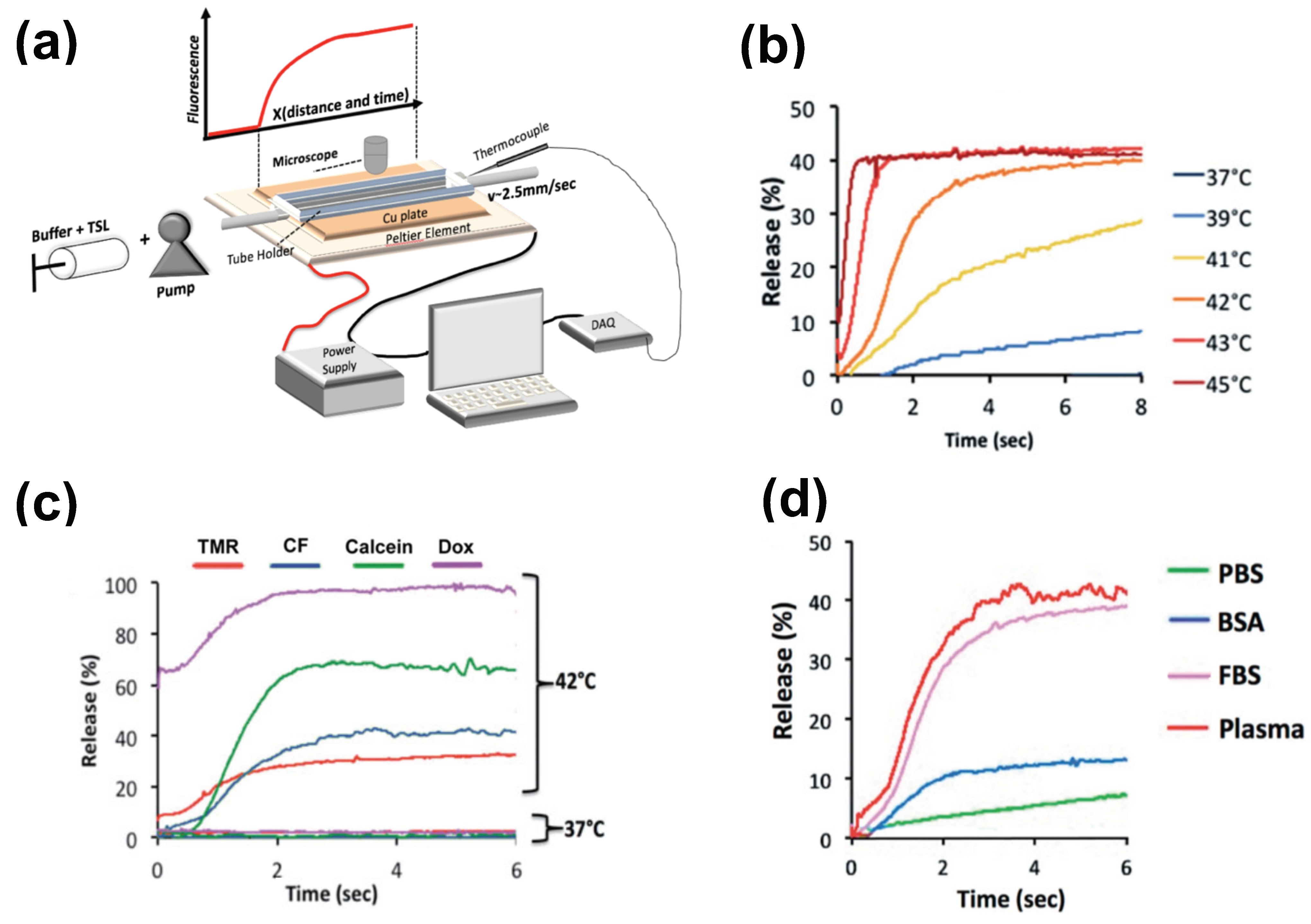
Figure 6. Measuring release kinetics of fast-release TSL formulations. (a) Millifluidic release assay schematics. A TSL solution (TSL + buffer) is pumped through a capillary tube that has been heated to the desired temperature by a Peltier element. Once the TSL solution enters the heated region, TSL begin to release the fluorescent drug/dye, resulting in a fluorescence gradient along the tube (upper graph). The Peltier temperature is measured by a thermocouple, and a control algorithm regulates the power applied to the Peltier element to control temperature. (b) Release of carboxyfluorescein (CF) from fast-releasing TSL (DPPC:MSPC:DSPE-PEG2000 = 85:10:5) between 37 and 45 °C during the first 8 s. Release within seconds is required to take advantage of the intra-vascular triggered release paradigm. (c) Release depends on the encapsulated compound, shown for four compounds for the same TSL formulation. Release in (a–c) was measured using fetal bovine serum (FBS) as buffer. (d) Release kinetics vary between buffers. CF release is shown for 4 buffers: phosphate buffered saline (PBS), 10% bovine serum albumin (BSA) solution, fetal bovine serum (FBS), and human plasma. TSL formulation used in (b–d) was identical. Figures reproduced with permission from [121].
2.1. TSL Release Kinetics
The early TSL formulations had comparably slow release (within minutes to hours) [122][123]. In addition, heating to >42 °C was required to achieve substantial release. This is disadvantageous since temperatures above 43 °C may result in reduced blood flow [124] that would also reduce the inflow of TSL-encapsulated drug. The first fast-release TSL formulations were published in the early 1990s, demonstrating substantial release within a few seconds after heating to >41 °C [125][126]. Such rapid release is required to take full advantage of the intravascular triggered release paradigm, as discussed above. The first fast-release formulation with substantial release at lower temperatures (40 °C) was presented around the year 2000 [31][127][128], and formed the basis for the first commercial TSL formulation (ThermoDox®) that has been employed in several human clinical trials [129][130][131][132][133][134][135]. Several additional fast-release TSL formulations encapsulating various agents have been presented within the last two decades [136][137][138]. Recent studies confirm that fast-release TSL that release within a few seconds can deliver substantially higher drug amounts compared to slower releasing formulations [20][21][22]. However, for most of these TSL formulations, release kinetics is not known within the time scale relevant for intravascular triggered release (e.g., within the first few seconds), owing to limitations of conventional methods used for measuring the release kinetics.
The most widely used method for measuring TSL release kinetics employs a buffer pre-heated to the desired temperature, where a small volume of TSL is added, typically under stirring [113][123][137][139][140][141][142][143]. Release is quantified usually using spectrophotometry, since optical properties (e.g., fluorescence) change when the drug is released from TSL. Due to the time required for mixing of the TSL with the buffer, the first reported time points are typically between 8–20 s. This time is substantially longer than many typical tumor transit times, making these measurements of limited value.
There have been two methods presented to measure TSL release kinetics at short (second) time scales. The first method employed a small-diameter tube within which TSL solution was passed through heated water for a specific time, and the released drug was quantified in the sample exiting the tube [125]. An advantage of this method is that various quantification methods can be employed on these samples. In a second method, a glass capillary tube was heated by a Peltier element, and release was quantified by measuring fluorescence along the tube by either microscopic or macroscopic fluorescence imaging (Figure 6a) [121][144]. This method provides data at high temporal resolution not possible with other methods (Figure 6b–d), but is limited to fluorescent agents. In both methods, it is important to select thin-walled tubes and to validate sufficiently rapid heating of the solution passing though the tube to target temperature [121].
To compare the release kinetics of TSL formulations, a recent study suggested using a characteristic release time based on a linear approximation of the TSL release kinetics [20]. As noted earlier, TSL only spend a few seconds within a tumor (=transit time), and for most TSL, the release kinetics within those first seconds can be adequately represented by a linear approximation (Figure 7). Ideally, this release time would be smaller than the transit time to maximize release and tissue drug uptake (Figure 5). The amount of drug released during tumor transit (Figure 5) can be estimated by the ratio of transit time to release time (can be found in original context). A recent study demonstrated that TSL with rapid release (i.e., short release time) can deliver substantially more drug to tissue than TSL with slow release (Figure 8) [20]. Table 1 summarizes the release times of published fast-release TSL formulations. In most cases, the exact release times could not be determined owing to limitations of methods used to quantify the release kinetics, as described above.
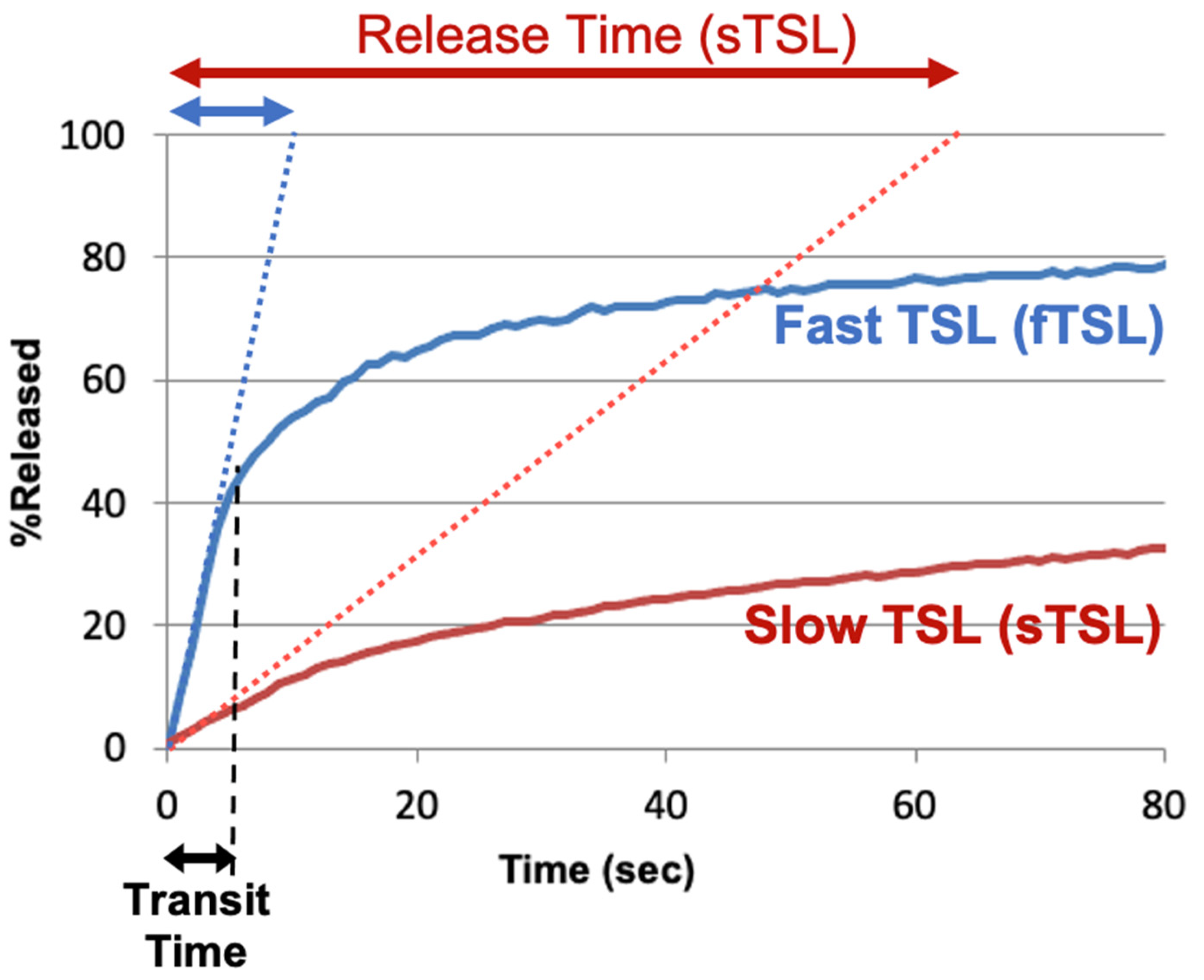
Figure 7. TSL release time. Release of two TSL formulations encapsulating a fluorescent drug analog (carboxyfluorescein) with slow (sTSL) and fast (fTSL) release is plotted, based on data from a prior study [20]. The dotted lines indicate a linear approximation of the release kinetics. TSL only spend a few seconds within the heated tumor (see black double arrow indicating ‘Transit Time’). Thus, in most cases, a linear approximation adequately represents release within those few seconds that TSL spend within the tumor vessels. Based on this linear approximation, a characteristic ‘release time’ is determined (indicated by red and blue double-arrows at the top) that enables the comparison of different TSL formulations. This release time was 8.2 s for fTSL, and 63.0 s for sTSL. The fraction of drug released during transit can be estimated by the ratio of transit time to release time (can be found in original context).
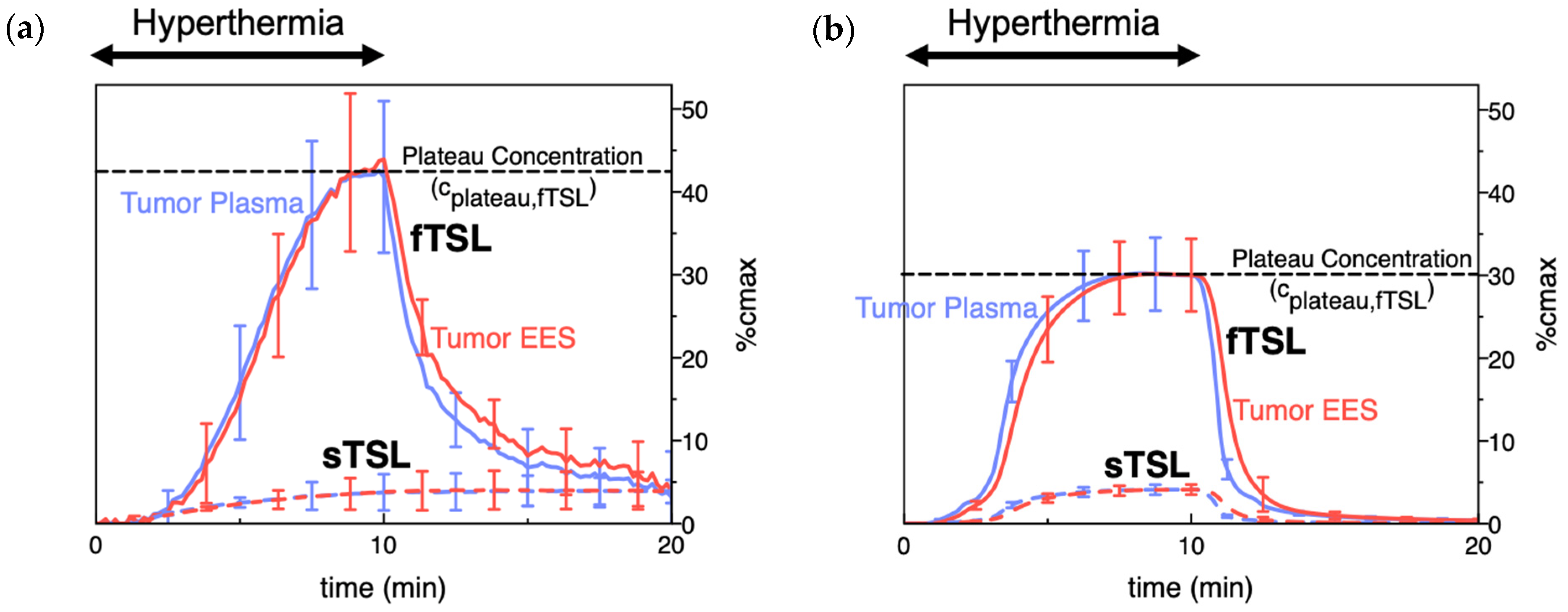
Figure 8. TSL delivery kinetics. (a) Drug concentration in plasma and interstitium (extracellular-extravascular space, EES) was determined from intravital microscopy data. Hyperthermia (42 °C) for 10 min was applied following the administration of either slow- (sTSL) or fast-release TSL (fTSL) encapsulating a fluorescent drug analog (carboxyfluorescein) (see Figure 7 for release kinetics of sTSL and fTSL). Plasma concentration increases during hyperthermia due to drug release. Released drug is then extracted by tissue, indicated by increasing interstitial (EES) concentration. A plateau (peak) concentration is approached towards the end of hyperthermia. This plateau concentration is substantially higher for fTSL compared to sTSL. Error bars indicate standard deviation (n = 3 animals/group). (b) Computer simulation of drug delivery kinetics based on in vivo measured tumor properties reproduces the delivery kinetics observed in (a). Error bars indicate computer model uncertainty due to uncertainty of model parameters. Figures reproduced from [20] (published under CC BY 4.0 license).
Table 1. Fast-release TSL formulations. Release times (see Figure 7) were estimated if possible, or an upper limit was provided (e.g., <20 s); in the latter cases, release at the first measured time point is shown in brackets. Buffer used for release measurement is indicated, since buffer affects release kinetics [121].
| TSL Composition (Molar Ratio) | Drug | Release Time [Temp.] | Buffer | In Vivo Plasma Half-Life (Species) | Refs. |
|---|---|---|---|---|---|
| DPPC:MSPC:DSPE-PEG2000 (86:10:4) | Doxorubicin | 3 s [40 °C] | human plasma | 0.96 h (human); 1–2 h (rabbit); 4.8 h (pig) |
[127][128][129][140][145][146][147] |
| DPPC:MSPC:DSPE-PEG2000 (85.3:9.7:5) | Doxorubicin | 4 s [41 °C] | PBS | 0.93 h (mouse); 0.96 h (rat); 0.75 h (dog) | [30][148][149] |
| DPPC:DSPC:DSPE-PEG2000 (70:25:5) | Doxorubicin | ~5–10 s [42 °C] | FBS | >1 h (mouse) | [105] |
| DPPC:DSPE-PEG2000:Ch:mELP | Doxorubicin | <5 s [41–42 °C] | FBS + culture media | 2.0 h (mouse) | [150] |
| DPPC:DSPC:DPPG1 (50:20:30) | Doxorubicin | <20 s [42 °C] (92.2% release @ 20 s) |
HEPES buffered saline | 1.4 h (rat) | [151] |
| DPPC:DSPC:DPPG2 (50:20:30) | Doxorubicin | <20 s [42 °C] (~75% release @ 20 s) |
HEPES buffered saline | ~1 h (pig); 1.6–2.4 h (rat); 0.4–0.7 h (cat) |
[151][152][153][154][155] |
| EYPC:Chol:Peg-PE:poly(EOEOVE-OD4) (50:45:4:2) | Doxorubicin | ~1 min [43 °C] | HEPES buffered saline | - | [156] |
| DPPC:Brij78 | Doxorubicin | ~1 min [42 °C] | FBS | 0.5 h (mouse) | [157] |
| DOPE:EPC:chol-pHPMAlac (70:25:5) | Doxorubicin | ~2 min [42 °C] | HEPES buffered saline | - | [158] |
| DPPC:DSPC:DSPE-PEG2000 (60:35:5) | Idarubicin | <1 s [42 °C] | FBS | >1 h (mouse) | [105] |
| DPPC:DSPC:DSPE-PEG2000 (80:15:5) | Gemcitabine | <2 min [42 °C] (90% release @ 2 min) |
FBS | ~2 h (mouse) | [143] |
| DPPC:MSPC:DSPE-PEG2000 (86:10:4) | Gemcitabine | ~30–60 s | FBS:saline (1:1) | - | [34] |
| DPPC:Brij78 | Gemcitabine | ~30–60 s | FBS:saline (1:1) | ~2 h (mouse) | [34] |
| DPPC:Brij78 | Oxiplatin | ~30–60 s | FBS:saline (1:1) | ~1 h (mouse) | [34] |
| DPPC:DSPC (90:10) | Cisplatin | 3–5 s [43 °C] | rat plasma | ~1 h (mouse) | [125][126][159] |
| DPPC:DPPG:MSPC:DSPE-PEG2000 (57.7:28.9:9.6:3.8) |
Cisplatin | <5 min [42 °C] (90% release @ 5 min) |
0.9% saline | ~1.5 h (mouse) | [113] |
| DPPC:MSPC:DSPG:DSPE-PEG2000 (82:8:10:4) | Epirubicin | ~4 min [41–43 °C] | PBS | 0.2 h (rat) | [160] |
| DPPC:MSPC:DSPE-PEG2000 (86:10:4) | Alvespimycin | <30 s [42 °C] (90% release @ 30 s) |
BSA in PBS | 0.2 h (mouse) | [80] |
DPPC: 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine; DSPC: 1,2-distearoyl-sn-glycero-3-phosphocholine; MSPC: 1-stearoyl-2-hydroxy-sn-glycero-3-phosphatidylcholine; DPPG: 1,2-dipalmitoyl- sn-glycero-3-phosphoglycerol; PE: poly ethylene; PEG: polyethylene glycol; Ch: Cholesterol; EYPC: egg yolk phosphatidylcholine; EOEOVE: 2-(2-ethoxy)ethoxyethyl vinyl ether; mELP: modified elastin-like polypeptide; Brij78: proprietary surfactant (main component: eicosaethylene glycol octadecyl ether); pHPMAlac: 2-Hydroxypropyl methacrylamide mono/dilactate polymers; PBS: phosphate buffered saline; FBS: fetal bovine serum; BSA: bovine serum albumin.
2.2. Plasma Stability
Plasma stability describes how long TSL-encapsulated drug remains in the systemic circulation after administration and can be quantified by the initial plasma half-life of a TSL formulation. Similar to the TSL release kinetics, plasma stability depends both on TSL formulation and encapsulated drug, but also varies with species (Table 1). During hyperthermia, circulating TSL-encapsulated drug continuously enters the heated tissue volume, with subsequent intravascular drug release (Figure 2 and Figure 5). The plasma concentration of TSL-encapsulated drug represents the amount available for intravascular triggered release. Thus, the AUC (area under the concentration vs. time curve) of the plasma concentration calculated during hyperthermia correlates with the total amount of TSL-encapsulated drug subjected to hyperthermia [161][162]. As a result, this AUC directly correlates with the amount of drug released in the heated tumor (can be found in original context). This AUC also correlates with tumor drug uptake, as initially demonstrated in a computer modeling study [161] and later confirmed by several experimental studies (Figure 9) [149][162][163]. A higher plasma stability would therefore increase this AUC, resulting in larger amount of drug being released—assuming that the kinetics of TSL release is not different (e.g., increased plasma stability of a TSL formulation may be disadvantageous if it is associated with slower release). Similarly, one approach to enhance drug delivery is to adjust the timing of hyperthermia as to maximize the plasma AUC during heating [161][163].
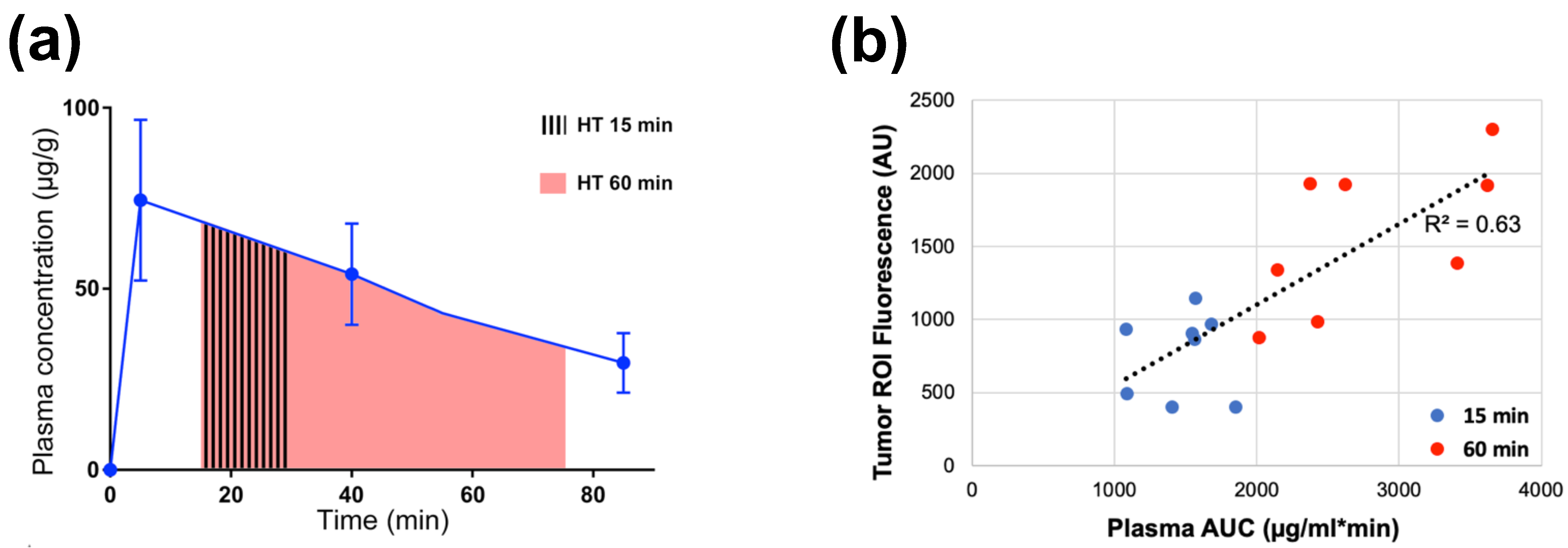
Figure 9. Plasma-AUC during hyperthermia correlates with tumor drug uptake. (a) AUC of plasma Dox concentration was calculated during heating, for either 15 or 60 min hyperthermia (HT) as indicated by shaded regions. (b) Plasma-AUC during HT correlated well with Dox fluorescence in the tumor region-of-interest measured following HT (R2 = 0.63). Tumors were exposed to hyperthermia (43 °C) for either 15 min (blue dots) or 60 min (red dots). Data reproduced from [162] (published under CC BY 4.0 license).
Note however that such comparisons based on AUC are only appropriate for different studies with the same TSL formulation, and the same or similar hyperthermia methods (i.e., with similar tumor temperature). The AUC indicates the total amount of TSL-encapsulated drug that passes through the heated tissue during hyperthermia. If two different heating devices with different temperature profiles and heating volumes are used, the amount of drug released from TSL will differ. Similarly, if two different TSL formulations are used, the amount of drug released will differ due to varying TSL release kinetics. Thus, even if the AUC is identical (=total amount of TSL-encapsulated drug passing through heated tissue), the amount released from these two TSL formulations will vary, resulting in different tumor drug uptake.
TSL plasma stability depends on several factors, and one major contributor is drug leakage from TSL at body temperature (37 °C)—i.e., drug slowly leaks from TSL while in systemic circulation [164]. Unfortunately, the release rate at body temperature is usually tied to the release rate at hyperthermic temperatures—i.e., slow release at 37 °C and rapid release at hyperthermia represent conflicting requirements for TSL formulations.
The peak plasma concentration after administration of TSL-encapsulated drug (Figure 9a) naturally correlates with the administered dose. Often, the administered dose is close to, or at the maximum tolerated dose (MTD) for that particular TSL–drug formulation in the studied species. In rodents, the MTD relative to body weight is often substantially higher compared to humans [165]. This higher administered dose in rodents results in higher plasma concentration (Figure 9a) and higher tumor drug uptake compared to large animals [147][149][155] and humans [129][133]. This issue may be relevant when extrapolating results on tumor drug uptake and therapeutic response from rodent studies to human patients.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15020398
References
- Kashkooli, F.M.; Soltani, M.; Souri, M. Controlled anti-cancer drug release through advanced nano-drug delivery systems: Static and dynamic targeting strategies. J. Control. Release 2020, 327, 316–349.
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003.
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827.
- Wang, Y.; Kohane, D.S. External triggering and triggered targeting strategies for drug delivery. Nat. Rev. Mater. 2017, 2, 17020.
- Yatvin, M.B.; Weinstein, J.N.; Dennis, W.H.; Blumenthal, R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science 1978, 202, 1290–1293.
- Weinstein, J.N.; Magin, R.L.; Yatvin, M.B.; Zaharko, D.S. Liposomes and local hyperthermia: Selective delivery of methotrexate to heated tumors. Science 1979, 204, 188–191.
- Yatvin, M.B.; Mühlensiepen, H.; Porschen, W.; Weinstein, J.N.; Feinendegen, L.E. Selective delivery of liposome-associated cis-dichlorodiammineplatinum(ii) by heat and its influence on tumor drug uptake and growth. Cancer Res. 1981, 41, 1602.
- Weinstein, J.N.; Magin, R.L.; Cysyk, R.L.; Zaharko, D.S. Treatment of solid l1210 murine tumors with local hyperthermia and temperature-sensitive liposomes containing methotrexate. Cancer Res. 1980, 40, 1388.
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: State of the art review. Int. J. Nanomed. 2014, 9, 4387–4398.
- Haemmerich, D.; Motamarry, A. Thermosensitive liposomes for image-guided drug delivery. In Advances in Cancer Research; Broome, A.-M., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 139, pp. 121–146.
- Aghdam, M.A.; Bagheri, R.; Mosafer, J.; Baradaran, B.; Hashemzaei, M.; Baghbanzadeh, A.; de la Guardia, M.; Mokhtarzadeh, A. Recent advances on thermosensitive and ph-sensitive liposomes employed in controlled release. J. Control. Release 2019, 315, 1–22.
- Al-Ahmady, Z.; Kostarelos, K. Chemical components for the design of temperature-responsive vesicles as cancer therapeutics. Chem. Rev. 2016, 116, 3883–3918.
- Mazzotta, E.; Tavano, L.; Muzzalupo, R. Thermo-sensitive vesicles in controlled drug delivery for chemotherapy. Pharmaceutics 2018, 10, 150.
- Seynhaeve, A.L.B.; Amin, M.; Haemmerich, D.; van Rhoon, G.C.; ten Hagen, T.L.M. Hyperthermia and smart drug delivery systems for solid tumor therapy. Adv. Drug Deliv. Rev. 2020, 163–164, 125–144.
- Ta, T.; Porter, T.M. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J. Control. Release Off. J. Control. Release Soc. 2013, 169, 112–125.
- Munaweera, I.; Shaikh, S.; Maples, D.; Nigatu, A.S.; Sethuraman, S.N.; Ranjan, A.; Greenberg, D.E.; Chopra, R. Temperature-sensitive liposomal ciprofloxacin for the treatment of biofilm on infected metal implants using alternating magnetic fields. Int. J. Hyperth. 2018, 34, 189–200.
- Wardlow, R.; Bing, C.; Van Osdol, J.; Maples, D.; Ladouceur-Wodzak, M.; Harbeson, M.; Nofiele, J.; Staruch, R.; Ramachandran, A.; Malayer, J.; et al. Targeted antibiotic delivery using low temperature-sensitive liposomes and magnetic resonance-guided high-intensity focused ultrasound hyperthermia. Int. J. Hyperth. 2016, 32, 254–264.
- Jeong, M.; Park, J.-H. Nanomedicine for the treatment of rheumatoid arthritis. Mol. Pharm. 2021, 18, 539–549.
- Saxena, V.; Johnson, C.G.; Negussie, A.H.; Sharma, K.V.; Dreher, M.R.; Wood, B.J. Temperature-sensitive liposome-mediated delivery of thrombolytic agents. Int. J. Hyperth. 2015, 31, 67–73.
- ten Hagen, T.L.M.; Dreher, M.R.; Zalba, S.; Seynhaeve, A.L.B.; Amin, M.; Li, L.; Haemmerich, D. Drug transport kinetics of intravascular triggered drug delivery systems. Commun. Biol. 2021, 4, 920.
- Manzoor, A.A.; Lindner, L.H.; Landon, C.D.; Park, J.-Y.; Simnick, A.J.; Dreher, M.R.; Das, S.; Hanna, G.; Park, W.; Chilkoti, A.; et al. Overcoming limitations in nanoparticle drug delivery: Triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012, 72, 5566–5575.
- Gasselhuber, A.; Dreher, M.R.; Rattay, F.; Wood, B.J.; Haemmerich, D. Comparison of conventional chemotherapy, stealth liposomes and temperature-sensitive liposomes in a mathematical model. PLoS One 2012, 7, e47453.
- Li, L.; Hagen, T.L.T.; Hossann, M.; Suss, R.; van Rhoon, G.C.; Eggermont, A.M.; Haemmerich, D.; Koning, G.A. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J. Control. Release Off. J. Control. Release Soc. 2013, 168, 142–150.
- Li, L.; Hagen, T.L.T.; Haeri, A.; Soullie, T.; Scholten, C.; Seynhaeve, A.L.; Eggermont, A.M.; Koning, G.A. A novel two-step mild hyperthermia for advanced liposomal chemotherapy. J. Control. Release Off. J. Control. Release Soc. 2013, 174, 202–208.
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer-chemotherapy—Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392.
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Dan, P. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410.
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014.
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37.
- Danhier, F. To exploit the tumor microenvironment: Since the epr effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121.
- Motamarry, A.; Negussie, A.H.; Rossmann, C.; Small, J.; Wolfe, A.M.; Wood, B.J.; Haemmerich, D. Real-time fluorescence imaging for visualization and drug uptake prediction during drug delivery by thermosensitive liposomes. Int. J. Hyperth. 2019, 36, 817–826.
- Kong, G.; Anyarambhatla, G.; Petros, W.P.; Braun, R.D.; Colvin, O.M.; Needham, D.; Dewhirst, M.W. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: Importance of triggered drug release. Cancer Res. 2000, 60, 6950–6957.
- Al-Jamal, W.T.; Kostarelos, K. Mild hyperthermia accelerates doxorubicin clearance from tumour-extravasated temperature-sensitive liposomes. Nanotheranostics 2022, 6, 230–242.
- Lokerse, W.J.; Bolkestein, M.; Ten Hagen, T.L.; de Jong, M.; Eggermont, A.M.; Grull, H.; Koning, G.A. Investigation of particle accumulation, chemosensitivity and thermosensitivity for effective solid tumor therapy using thermosensitive liposomes and hyperthermia. Theranostics 2016, 6, 1717–1731.
- May, J.P.; Ernsting, M.J.; Undzys, E.; Li, S.-D. Thermosensitive liposomes for the delivery of gemcitabine and oxaliplatin to tumors. Mol. Pharm. 2013, 10, 4499–4508.
- Chen, Y.; Xia, R.; Huang, Y.; Zhao, W.; Li, J.; Zhang, X.; Wang, P.; Venkataramanan, R.; Fan, J.; Xie, W.; et al. An immunostimulatory dual-functional nanocarrier that improves cancer immunochemotherapy. Nat. Commun. 2016, 7, 13443.
- Dai, Y.; Xing, H.; Song, F.; Yang, Y.; Qiu, Z.; Lu, X.; Liu, Q.; Ren, S.; Chen, X.; Li, N. Biotin-conjugated multilayer poly -lecithin-polyethylene glycol nanoparticles for targeted delivery of doxorubicin. J. Pharm. Sci. 2016, 105, 2949–2958.
- He, C.; Poon, C.; Chan, C.; Yamada, S.D.; Lin, W. Nanoscale coordination polymers codeliver chemotherapeutics and sirnas to eradicate tumors of cisplatin-resistant ovarian cancer. J. Am. Chem. Soc. 2016, 138, 6010–6019.
- Huang, P.; Liu, J.; Wang, W.; Zhang, Y.; Zhao, F.; Kong, D.; Liu, J.; Dong, A. Zwitterionic nanoparticles constructed from bioreducible raft–rop double head agent for shell shedding triggered intracellular drug delivery. Acta. Biomater. 2016, 40, 263–272.
- Li, Z.; Hu, Y.; Howard, K.A.; Jiang, T.; Fan, X.; Miao, Z.; Sun, Y.; Besenbacher, F.; Yu, M. Multifunctional bismuth selenide nanocomposites for antitumor thermo-chemotherapy and imaging. ACS Nano 2016, 10, 984–997.
- Liu, H.; Gao, M.; Xu, H.; Guan, X.; Lv, L.; Deng, S.; Zhang, C.; Tian, Y. A promising emodin-loaded poly (lactic-co-glycolic acid)-d-α-tocopheryl polyethylene glycol 1000 succinate nanoparticles for liver cancer therapy. Pharm. Res. 2016, 33, 217–236.
- Liu, L.X.; Li, B.X.; Wang, Q.Y.; Dong, Z.P.; Li, H.M.; Jin, Q.M.; Hong, H.; Zhang, J.; Wang, Y. An integrative folate-based metal complex nanotube as a potent antitumor nanomedicine as well as an efficient tumor-targeted drug carrier. Bioconjugate Chem. 2016, 27, 2863–2873.
- Mei, L.; Liu, Y.; Zhang, H.; Zhang, Z.; Gao, H.; He, Q. Antitumor and antimetastasis activities of heparin-based micelle served as both carrier and drug. ACS Appl. Mater. Interfaces 2016, 8, 9577–9589.
- Nascimento, A.V.; Gattacceca, F.; Singh, A.; Bousbaa, H.; Ferreira, D.; Sarmento, B.; Amiji, M.M. Biodistribution and pharmacokinetics of mad2 sirna-loaded egfr-targeted chitosan nanoparticles in cisplatin sensitive and resistant lung cancer models. Nanomed. (Lond) 2016, 11, 767–781.
- Shi, J.; Chen, Z.; Wang, L.; Wang, B.; Xu, L.; Hou, L.; Zhang, Z. A tumor-specific cleavable nanosystem of peg-modified hybrid aggregates for radio frequency-controlled release, hyperthermia, photodynamic therapy and x-ray imaging. Acta. Biomater. 2016, 29, 282–297.
- Tang, Z.; Zhang, L.; Wang, Y.; Li, D.; Zhong, Z.; Zhou, S. Redox-responsive star-shaped magnetic micelles with active-targeted and magnetic-guided functions for cancer therapy. Acta. Biomater. 2016, 42, 232–246.
- Tomalova, B.; Sirova, M.; Rossmann, P.; Pola, R.; Strohalm, J.; Chytil, P.; Cerny, V.; Tomala, J.; Kabesova, M.; Rihova, B.; et al. The structure-dependent toxicity, pharmacokinetics and anti-tumour activity of hpma copolymer conjugates in the treatment of solid tumours and leukaemia. J. Control. Release 2016, 223, 1–10.
- Zhang, L.; Li, G.; Gao, M.; Liu, X.; Ji, B.; Hua, R.; Zhou, Y.; Yang, Y. Rgd-peptide conjugated inulin-ibuprofen nanoparticles for targeted delivery of epirubicin. Colloids Surf. B Biointerfaces 2016, 144, 81–89.
- Zhang, R.X.; Cai, P.; Zhang, T.; Chen, K.; Li, J.; Cheng, J.; Pang, K.S.; Adissu, H.A.; Rauth, A.M.; Wu, X.Y. Polymer–lipid hybrid nanoparticles synchronize pharmacokinetics of co-encapsulated doxorubicin–mitomycin c and enable their spatiotemporal co-delivery and local bioavailability in breast tumor. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1279–1290.
- Zou, Y.; Fang, Y.; Meng, H.; Meng, F.; Deng, C.; Zhang, J.; Zhong, Z. Self-crosslinkable and intracellularly decrosslinkable biodegradable micellar nanoparticles: A robust, simple and multifunctional nanoplatform for high-efficiency targeted cancer chemotherapy. J. Control. Release 2016, 244, 326–335.
- Boissenot, T.; Bordat, A.; Larrat, B.; Varna, M.; Chacun, H.; Paci, A.; Poinsignon, V.; Fattal, E.; Tsapis, N. Ultrasound-induced mild hyperthermia improves the anticancer efficacy of both taxol® and paclitaxel-loaded nanocapsules. J. Control. Release 2017, 264, 219–227.
- Deng, H.; Zhao, X.; Deng, L.; Liu, J.; Dong, A. Reactive oxygen species activated nanoparticles with tumor acidity internalization for precise anticancer therapy. J. Control. Release 2017, 255, 142–153.
- Gaonkar, R.H.; Ganguly, S.; Dewanjee, S.; Sinha, S.; Gupta, A.; Ganguly, S.; Chattopadhyay, D.; Chatterjee Debnath, M. Garcinol loaded vitamin e tpgs emulsified plga nanoparticles: Preparation, physicochemical characterization, in vitro and in vivo studies. Sci. Rep. 2017, 7, 530.
- Gou, J.; Liang, Y.; Miao, L.; Guo, W.; Chao, Y.; He, H.; Zhang, Y.; Yang, J.; Wu, C.; Yin, T.; et al. Improved tumor tissue penetration and tumor cell uptake achieved by delayed charge reversal nanoparticles. Acta. Biomater. 2017, 62, 157–166.
- He, R.; Yin, C. Trimethyl chitosan based conjugates for oral and intravenous delivery of paclitaxel. Acta. Biomater. 2017, 53, 355–366.
- Hou, J.; Guo, C.; Shi, Y.; Liu, E.; Dong, W.; Yu, B.; Liu, S.; Gong, J. A novel high drug loading mussel-inspired polydopamine hybrid nanoparticle as a ph-sensitive vehicle for drug delivery. Int. J. Pharm. 2017, 533, 73–83.
- Hou, L.; Shan, X.; Hao, L.; Feng, Q.; Zhang, Z. Copper sulfide nanoparticle-based localized drug delivery system as an effective cancer synergistic treatment and theranostic platform. Acta. Biomater. 2017, 54, 307–320.
- Huo, M.; Wang, L.; Chen, Y.; Shi, J. Tumor-selective catalytic nanomedicine by nanocatalyst delivery. Nat. Commun. 2017, 8, 357.
- Kong, M.; Tang, J.; Qiao, Q.; Wu, T.; Qi, Y.; Tan, S.; Gao, X.; Zhang, Z. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics 2017, 7, 3276–3292.
- Logie, J.; Ganesh, A.N.; Aman, A.M.; Al-awar, R.S.; Shoichet, M.S. Preclinical evaluation of taxane-binding peptide-modified polymeric micelles loaded with docetaxel in an orthotopic breast cancer mouse model. Biomaterials 2017, 123, 39–47.
- Roy, A.; Zhao, Y.; Yang, Y.; Szeitz, A.; Klassen, T.; Li, S.-D. Selective targeting and therapy of metastatic and multidrug resistant tumors using a long circulating podophyllotoxin nanoparticle. Biomaterials 2017, 137, 11–22.
- Shalgunov, V.; Zaytseva-Zotova, D.; Zintchenko, A.; Levada, T.; Shilov, Y.; Andreyev, D.; Dzhumashev, D.; Metelkin, E.; Urusova, A.; Demin, O.; et al. Comprehensive study of the drug delivery properties of poly(l-lactide)-poly(ethylene glycol) nanoparticles in rats and tumor-bearing mice. J. Control. Release 2017, 261, 31–42.
- Su, J.; Sun, H.; Meng, Q.; Zhang, P.; Yin, Q.; Li, Y. Enhanced blood suspensibility and laser-activated tumor-specific drug release of theranostic mesoporous silica nanoparticles by functionalizing with erythrocyte membranes. Theranostics 2017, 7, 523–537.
- Wang, J.; Lee, G.Y.; Lu, Q.; Peng, X.; Wu, J.; Wu, S.; Kairdolf, B.A.; Nie, S.; Wang, Y.; Lane, L.A. Quantitative examination of the active targeting effect: The key factor for maximal tumor accumulation and retention of short-circulated biopolymeric nanocarriers. Bioconjugate Chem. 2017, 28, 1351–1355.
- Wang, L.; Li, D.; Hao, Y.; Niu, M.; Hu, Y.; Zhao, H.; Chang, J.; Zhang, Z.; Zhang, Y. Gold nanorod-based poly(lactic-co-glycolic acid) with manganese dioxide core-shell structured multifunctional nanoplatform for cancer theranostic applications. Int. J. Nanomed. 2017, 12, 3059–3075.
- Wu, J.; Tang, C.; Yin, C. Co-delivery of doxorubicin and interleukin-2 via chitosan based nanoparticles for enhanced antitumor efficacy. Acta. Biomater. 2017, 47, 81–90.
- Xue, H.; Yu, Z.; Liu, Y.; Yuan, W.; Yang, T.; You, J.; He, X.; Lee, R.J.; Li, L.; Xu, C. Delivery of mir-375 and doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple drug resistance in hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 5271–5287.
- Yan, G.; Wang, J.; Hu, L.; Wang, X.; Yang, G.; Fu, S.; Cheng, X.; Zhang, P.; Tang, R. Stepwise targeted drug delivery to liver cancer cells for enhanced therapeutic efficacy by galactose-grafted, ultra-ph-sensitive micelles. Acta. Biomater. 2017, 51, 363–373.
- Yan, G.; Wang, J.; Qin, J.; Hu, L.; Zhang, P.; Wang, X.; Tang, R. Well-defined poly(ortho ester amides) for potential drug carriers: Probing the effect of extra- and intracellular drug release on chemotherapeutic efficacy. Macromol. Biosci. 2017, 17, 1600503.
- Ansari, L.; Jaafari, M.R.; Bastami, T.R.; Malaekeh-Nikouei, B. Improved anticancer efficacy of epirubicin by magnetic mesoporous silica nanoparticles: In vitro and in vivo studies. Artif. Cells Nanomed. Biotechnol. 2018, 46, 594–606.
- Chai, S.; Kan, S.; Sun, R.; Zhou, R.; Sun, Y.; Chen, W.; Yu, B. Fabricating polydopamine-coated mose2-wrapped hollow mesoporous silica nanoplatform for controlled drug release and chemo-photothermal therapy. Int. J. Nanomed. 2018, 13, 7607–7621.
- Cheng, X.; Li, D.; Lin, A.; Xu, J.; Wu, L.; Gu, H.; Huang, Z.; Liu, J.; Zhang, Y.; Yin, X. Fabrication of multifunctional triple-responsive platform based on cus-capped periodic mesoporous organosilica nanoparticles for chemo-photothermal therapy. Int. J. Nanomed. 2018, 13, 3661–3677.
- Duan, D.; Wang, A.; Ni, L.; Zhang, L.; Yan, X.; Jiang, Y.; Mu, H.; Wu, Z.; Sun, K.; Li, Y. Trastuzumab- and fab’ fragment-modified curcumin peg-plga nanoparticles: Preparation and evaluation in vitro and in vivo. Int. J. Nanomed. 2018, 13, 1831–1840.
- Park, J.; Park, J.E.; Hedrick, V.E.; Wood, K.V.; Bonham, C.; Lee, W.; Yeo, Y. A comparative in vivo study of albumin-coated paclitaxel nanocrystals and abraxane. Small 2018, 14, e1703670.
- Srimathveeravalli, G.; Abdel-Atti, D.; Pérez-Medina, C.; Takaki, H.; Solomon, S.B.; Mulder, W.J.M.; Reiner, T. Reversible electroporation–mediated liposomal doxorubicin delivery to tumors can be monitored with 89zr-labeled reporter nanoparticles. Mol. Imaging 2018, 17, 1536012117749726.
- Wu, F.; Zhang, M.; Lu, H.; Liang, D.; Huang, Y.; Xia, Y.; Hu, Y.; Hu, S.; Wang, J.; Yi, X.; et al. Triple stimuli-responsive magnetic hollow porous carbon-based nanodrug delivery system for magnetic resonance imaging-guided synergistic photothermal/chemotherapy of cancer. ACS Appl. Mater. Interfaces 2018, 10, 21939–21949.
- Xu, C.; Chen, F.; Valdovinos, H.F.; Jiang, D.; Goel, S.; Yu, B.; Sun, H.; Barnhart, T.E.; Moon, J.J.; Cai, W. Bacteria-like mesoporous silica-coated gold nanorods for positron emission tomography and photoacoustic imaging-guided chemo-photothermal combined therapy. Biomaterials 2018, 165, 56–65.
- Yu, G.; Yang, Z.; Fu, X.; Yung, B.C.; Yang, J.; Mao, Z.; Shao, L.; Hua, B.; Liu, Y.; Zhang, F.; et al. Polyrotaxane-based supramolecular theranostics. Nat. Commun. 2018, 9, 766.
- Zhang, Y.; Guo, Z.; Cao, Z.; Zhou, W.; Zhang, Y.; Chen, Q.; Lu, Y.; Chen, X.; Guo, Q.; Li, C.; et al. Endogenous albumin-mediated delivery of redox-responsive paclitaxel-loaded micelles for targeted cancer therapy. Biomaterials 2018, 183, 243–257.
- Duan, X.; Chan, C.; Han, W.; Guo, N.; Weichselbaum, R.R.; Lin, W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat. Commun. 2019, 10, 1899.
- Dunne, M.; Epp-Ducharme, B.; Sofias, A.M.; Regenold, M.; Dubins, D.N.; Allen, C. Heat-activated drug delivery increases tumor accumulation of synergistic chemotherapies. J. Control. Release 2019, 308, 197–208.
- Jadon, R.S.; Sharma, M. Docetaxel-loaded lipid-polymer hybrid nanoparticles for breast cancer therapeutics. J. Drug Deliv. Sci. Technol. 2019, 51, 475–484.
- Mondal, L.; Mukherjee, B.; Das, K.; Bhattacharya, S.; Dutta, D.; Chakraborty, S.; Pal, M.M.; Gaonkar, R.H.; Debnath, M.C. Cd-340 functionalized doxorubicin-loaded nanoparticle induces apoptosis and reduces tumor volume along with drug-related cardiotoxicity in mice. Int. J. Nanomed. 2019, 14, 8073–8094.
- Mukerabigwi, J.F.; Yin, W.; Zha, Z.; Ke, W.; Wang, Y.; Chen, W.; Japir, A.A.-W.M.M.; Wang, Y.; Ge, Z. Polymersome nanoreactors with tumor ph-triggered selective membrane permeability for prodrug delivery, activation, and combined oxidation-chemotherapy. J. Control. Release 2019, 303, 209–222.
- Parker, C.L.; McSweeney, M.D.; Lucas, A.T.; Jacobs, T.M.; Wadsworth, D.; Zamboni, W.C.; Lai, S.K. Pretargeted delivery of peg-coated drug carriers to breast tumors using multivalent, bispecific antibody against polyethylene glycol and her2. Nanomed. Nanotechnol. Biol. Med. 2019, 21, 102076.
- Šimek, M.; Hermannová, M.; Šmejkalová, D.; Foglová, T.; Souček, K.; Binó, L.; Velebný, V. Lc–ms/ms study of in vivo fate of hyaluronan polymeric micelles carrying doxorubicin. Carbohydr. Polym. 2019, 209, 181–189.
- Wang, Y.; Liu, H.; Yao, D.; Li, J.; Yang, S.; Zhang, C.; Chen, W.; Wang, D. 18f-labeled magnetic nanoparticles for monitoring anti-angiogenic therapeutic effects in breast cancer xenografts. J. Nanobiotechnology 2019, 17, 105.
- Zhang, L.; Su, H.; Wang, H.; Li, Q.; Li, X.; Zhou, C.; Xu, J.; Chai, Y.; Liang, X.; Xiong, L.; et al. Tumor chemo-radiotherapy with rod-shaped and spherical gold nano probes: Shape and active targeting both matter. Theranostics 2019, 9, 1893–1908.
- Zhang, Y.; Liu, Y.; Gao, X.; Li, X.; Niu, X.; Yuan, Z.; Wang, W. Near-infrared-light induced nanoparticles with enhanced tumor tissue penetration and intelligent drug release. Acta. Biomater. 2019, 90, 314–323.
- Bort, G.; Lux, F.; Dufort, S.; Crémillieux, Y.; Verry, C.; Tillement, O. Epr-mediated tumor targeting using ultrasmall-hybrid nanoparticles: From animal to human with theranostic aguix nanoparticles. Theranostics 2020, 10, 1319–1331.
- Cong, Z.; Zhang, L.; Ma, S.-Q.; Lam, K.S.; Yang, F.-F.; Liao, Y.-H. Size-transformable hyaluronan stacked self-assembling peptide nanoparticles for improved transcellular tumor penetration and photo–chemo combination therapy. ACS Nano 2020, 14, 1958–1970.
- Ding, Y.; Sun, Z.; Tong, Z.; Zhang, S.; Min, J.; Xu, Q.; Zhou, L.; Mao, Z.; Xia, H.; Wang, W. Tumor microenvironment-responsive multifunctional peptide coated ultrasmall gold nanoparticles and their application in cancer radiotherapy. Theranostics 2020, 10, 5195–5208.
- Guo, J.; Yu, Z.; Das, M.; Huang, L. Nano codelivery of oxaliplatin and folinic acid achieves synergistic chemo-immunotherapy with 5-fluorouracil for colorectal cancer and liver metastasis. ACS Nano 2020, 14, 5075–5089.
- Hao, Q.; Wang, Z.; Zhao, W.; Wen, L.; Wang, W.; Lu, S.; Xing, D.; Zhan, M.; Hu, X. Dual-responsive polyprodrug nanoparticles with cascade-enhanced magnetic resonance signals for deep-penetration drug release in tumor therapy. ACS Appl. Mater. Interfaces 2020, 12, 49489–49501.
- Katifelis, H.; Mukha, I.; Bouziotis, P.; Vityuk, N.; Tsoukalas, C.; Lazaris, A.C.; Lyberopoulou, A.; Theodoropoulos, G.E.; Efstathopoulos, E.P.; Gazouli, M. Ag/au bimetallic nanoparticles inhibit tumor growth and prevent metastasis in a mouse model. Int. J. Nanomed. 2020, 15, 6019–6032.
- Kazi, J.; Sen, R.; Ganguly, S.; Jha, T.; Ganguly, S.; Chatterjee Debnath, M. Folate decorated epigallocatechin-3-gallate (egcg) loaded plga nanoparticles; in-vitro and in-vivo targeting efficacy against mda-mb-231 tumor xenograft. Int. J. Pharm. 2020, 585, 119449.
- Mu, J.; Zhong, H.; Zou, H.; Liu, T.; Yu, N.; Zhang, X.; Xu, Z.; Chen, Z.; Guo, S. Acid-sensitive pegylated paclitaxel prodrug nanoparticles for cancer therapy: Effect of peg length on antitumor efficacy. J. Control. Release 2020, 326, 265–275.
- Owen, J.; Thomas, E.; Menon, J.; Gray, M.; Skaripa-Koukelli, I.; Gill, M.R.; Wallington, S.; Miller, R.L.; Vallis, K.A.; Carlisle, R. Indium-111 labelling of liposomal hegf for radionuclide delivery via ultrasound-induced cavitation. J. Control. Release 2020, 319, 222–233.
- Sofias, A.M.; Toner, Y.C.; Meerwaldt, A.E.; van Leent, M.M.T.; Soultanidis, G.; Elschot, M.; Gonai, H.; Grendstad, K.; Flobak, Å.; Neckmann, U.; et al. Tumor targeting by αvβ3-integrin-specific lipid nanoparticles occurs via phagocyte hitchhiking. ACS Nano 2020, 14, 7832–7846.
- Xie, B.; Wan, J.; Chen, X.; Han, W.; Wang, H. Preclinical evaluation of a cabazitaxel prodrug using nanoparticle delivery for the treatment of taxane-resistant malignancies. Mol. Cancer Ther. 2020, 19, 822–834.
- Xiong, X.; Xu, Z.; Huang, H.; Wang, Y.; Zhao, J.; Guo, X.; Zhou, S. A nir light triggered disintegratable nanoplatform for enhanced penetration and chemotherapy in deep tumor tissues. Biomaterials 2020, 245, 119840.
- Zhou, Z.; Zhang, Q.; Yang, R.; Wu, H.; Zhang, M.; Qian, C.; Chen, X.; Sun, M. Atp-charged nanoclusters enable intracellular protein delivery and activity modulation for cancer theranostics. iScience 2020, 23, 100872.
- Cvjetinović, Đ.; Prijović, Ž.; Janković, D.; Radović, M.; Mirković, M.; Milanović, Z.; Mojović, M.; Škalamera, Đ.; Vranješ-Đurić, S. Bioevaluation of glucose-modified liposomes as a potential drug delivery system for cancer treatment using 177-lu radiotracking. J. Control. Release 2021, 332, 301–311.
- El-Safoury, D.M.; Ibrahim, A.B.; El-Setouhy, D.A.; Khowessah, O.M.; Motaleb, M.A.; Sakr, T.M. Amelioration of tumor targeting and in vivo biodistribution of 99mtc-methotrexate-gold nanoparticles (99mtc-mex-aunps). J. Pharm. Sci. 2021, 110, 2955–2965.
- El-Safoury, D.M.; Ibrahim, A.B.; El-Setouhy, D.A.; Khowessah, O.M.; Motaleb, M.A.; Sakr, T.M. Gold nanoparticles for 99mtc-doxorubicin delivery: Formulation, in vitro characterization, comparative studies in vivo stability and biodistribution. J. Radioanal. Nucl. Chem. 2021, 328, 325–338.
- Lu, T.; Haemmerich, D.; Liu, H.; Seynhaeve, A.L.B.; van Rhoon, G.C.; Houtsmuller, A.B.; ten Hagen, T.L.M. Externally triggered smart drug delivery system encapsulating idarubicin shows superior kinetics and enhances tumoral drug uptake and response. Theranostics 2021, 11, 5700–5712.
- Nabi, P.N.; Vahidfar, N.; Tohidkia, M.R.; Hamidi, A.A.; Omidi, Y.; Aghanejad, A. Mucin-1 conjugated polyamidoamine-based nanoparticles for image-guided delivery of gefitinib to breast cancer. Int. J. Biol. Macromol. 2021, 174, 185–197.
- Parakhonskiy, B.V.; Shilyagina, N.Y.; Gusliakova, O.I.; Volovetskiy, A.B.; Kostyuk, A.B.; Balalaeva, I.V.; Klapshina, L.G.; Lermontova, S.A.; Tolmachev, V.; Orlova, A.; et al. A method of drug delivery to tumors based on rapidly biodegradable drug-loaded containers. Appl. Mater. Today 2021, 25, 101199.
- Han, Y.; Dong, Z.; Wang, C.; Li, Q.; Hao, Y.; Yang, Z.; Zhu, W.; Zhang, Y.; Liu, Z.; Feng, L. Ferrous ions doped calcium carbonate nanoparticles potentiate chemotherapy by inducing ferroptosis. J. Control. Release 2022, 348, 346–356.
- Kannaka, K.; Sano, K.; Munekane, M.; Yamasaki, T.; Hagimori, M.; Mukai, T. Enhanced therapeutic effect of liposomal doxorubicin via bio-orthogonal chemical reactions in tumors. Mol. Pharm. 2022, 19, 1400–1409.
- Liping, Y.; Jian, H.; Zhenchao, T.; Yan, Z.; Jing, Y.; Yangyang, Z.; Jing, G.; Liting, Q. Gsh-responsive poly-resveratrol based nanoparticles for effective drug delivery and reversing multidrug resistance. Drug Deliv. 2022, 29, 229–237.
- Yarmolenko, P.S.; Zhao, Y.; Landon, C.; Spasojevic, I.; Yuan, F.; Needham, D.; Viglianti, B.L.; Dewhirst, M.W. Comparative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumours. Int. J. Hyperth. 2010, 26, 485–498.
- Schmidt, R. Neuartige Thermosensitive Liposomen Zur Zielgerichteten Therapie Solider Tumoren; Ludwig-Maximilians-Universität München: München, Germany, 2011.
- Dou, Y.N.; Zheng, J.; Foltz, W.D.; Weersink, R.; Chaudary, N.; Jaffray, D.A.; Allen, C. Heat-activated thermosensitive liposomal cisplatin (htlc) results in effective growth delay of cervical carcinoma in mice. J. Control. Release 2014, 178, 69–78.
- Dou, Y.N.; Chaudary, N.; Chang, M.C.; Dunne, M.; Huang, H.; Jaffray, D.A.; Milosevic, M.; Allen, C. Tumor microenvironment determines response to a heat-activated thermosensitive liposome formulation of cisplatin in cervical carcinoma. J. Control. Release Off. J. Control. Release Soc. 2017, 262, 182–191.
- Øye, K.S.; Gulati, G.; Graff, B.A.; Gaustad, J.-V.; Brurberg, K.G.; Rofstad, E.K. A novel method for mapping the heterogeneity in blood supply to normal and malignant tissues in the mouse dorsal window chamber. Microvasc. Res. 2008, 75, 179–187.
- Abdullah, S.S.; Pialat, J.B.; Wiart, M.; Duboeuf, F.; Mabrut, J.Y.; Bancel, B.; Rode, A.; Ducerf, C.; Baulieux, J.; Berthezene, Y. Characterization of hepatocellular carcinoma and colorectal liver metastasis by means of perfusion mri. J. Magn. Reson. Imaging 2008, 28, 390–395.
- Ludemann, L.; Prochnow, D.; Rohlfing, T.; Franiel, T.; Warmuth, C.; Taupitz, M.; Rehbein, H.; Beyersdorff, D. Simultaneous quantification of perfusion and permeability in the prostate using dynamic contrast-enhanced magnetic resonance imaging with an inversion-prepared dual-contrast sequence. Ann. Biomed. Eng. 2009, 37, 749–762.
- Rumboldt, Z.; Al-Okaili, R.; Deveikis, J.P. Perfusion ct for head and neck tumors: Pilot study. AJNR Am. J. Neuroradiol 2005, 26, 1178–1185.
- Notohamiprodjo, M.; Sourbron, S.; Staehler, M.; Michaely, H.J.; Attenberger, U.I.; Schmidt, G.P.; Boehm, H.; Horng, A.; Glaser, C.; Stief, C.; et al. Measuring perfusion and permeability in renal cell carcinoma with dynamic contrast-enhanced mri: A pilot study. J. Magn. Reson. Imaging 2010, 31, 490–501.
- Brix, G.; Kiessling, F.; Lucht, R.; Darai, S.; Wasser, K.; Delorme, S.; Griebel, J. Microcirculation and microvasculature in breast tumors: Pharmacokinetic analysis of dynamic mr image series. Magn. Reson. Med. 2004, 52, 420–429.
- Burke, C.; Dreher, M.R.; Negussie, A.H.; Mikhail, A.S.; Yarmolenko, P.; Patel, A.; Skilskyj, B.; Wood, B.J.; Haemmerich, D. Drug release kinetics of temperature sensitive liposomes measured at high-temporal resolution with a millifluidic device. Int. J. Hyperth. 2018, 34, 786–794.
- Magin, R.L.; Hunter, J.M.; Niesman, M.R.; Bark, G.A. Effect of vesicle size on the clearance, distribution, and tumor uptake of temperature-sensitive liposomes. Cancer Drug Deliv. 1986, 3, 223–237.
- Needham, D.; Dewhirst, M.W. The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Adv. Drug Deliv. Rev. 2001, 53, 285–305.
- Rossmann, C.; Haemmerich, D. Review of temperature dependence of thermal properties, dielectric properties, and perfusion of biological tissues at hyperthermic and ablation temperatures. Crit. Rev. Biomed. Eng. 2014, 42, 467–492.
- Iga, K.; Hamaguchi, N.; Igari, Y.; Ogawa, Y.; Gotoh, K.; Ootsu, K.; Toguchi, H.; Shimamoto, T. Enhanced antitumor activity in mice after administration of thermosensitive liposome encapsulating cisplatin with hyperthermia. J. Pharm. Exp. Ther. 1991, 257, 1203–1207.
- Iga, K. Optimum formulation of thermosensitive liposome for targeted tumor drug delivery. J. Takeda. Res. Lab. 1992, 51, 45–72.
- Anyarambhatla, G.R.; Needham, D. Enhancement of the phase transition permeability of dppc liposomes by incorporation of mppc: A new temperature-sensitive liposome for use with mild hyperthermia. J. Liposome Res. 1999, 9, 491–506.
- Needham, D.; Anyarambhatla, G.; Kong, G.; Dewhirst, M.W. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Res. 2000, 60, 1197–1201.
- Wood, B.J.; Poon, R.T.; Locklin, J.K.; Dreher, M.R.; Ng, K.K.; Eugeni, M.; Seidel, G.; Dromi, S.; Neeman, Z.; Kolf, M.; et al. Phase i study of heat-deployed liposomal doxorubicin during radiofrequency ablation for hepatic malignancies. J. Vasc. Interv. Radiol. 2012, 23, 248–255.
- Lencioni, R.; Tak, W.-Y.; Chen, M.H.; Finn, R.S.; Sherman, M.; Makris, L.; O’Neal, M.; Simonich, W.; Haemmerich, D.; Reed, R.; et al. Standardized radiofrequency ablation (srfa) ≥ 45 minutes (m) plus lyso-thermosensitive liposomal doxorubicin (ltld) for solitary hepatocellular carcinoma (hcc) lesions 3-7 cm: A retrospective analysis of phase iii heat study. J. Clin. Oncol. 2014, 32, e15143.
- Zagar, T.M.; Vujaskovic, Z.; Formenti, S.; Rugo, H.; Muggia, F.; O’Connor, B.; Myerson, R.; Stauffer, P.; Hsu, I.C.; Diederich, C.; et al. Two phase i dose-escalation/pharmacokinetics studies of low temperature liposomal doxorubicin (ltld) and mild local hyperthermia in heavily pretreated patients with local regionally recurrent breast cancer. Int. J. Hyperth. 2014, 30, 285–294.
- Kim, A.; Sharma, K.; Yarmolenko, P.; Celik, H.; Kaplan, R.N.; Dome, J.; Musso, L.; Borys, N.; Partanen, A.; Warner, L.; et al. Phase 1 trial of lyso-thermosensitive liposomal doxorubicin (ltld) and magnetic resonance guided high intensity focused ultrasound (mr-hifu) for pediatric refractory solid tumors. J. Clin. Oncol. 2017, 35, TPS10579.
- Lyon, P.C.; Gray, M.D.; Mannaris, C.; Folkes, L.K.; Stratford, M.; Campo, L.; Chung, D.Y.F.; Scott, S.; Anderson, M.; Goldin, R.; et al. Safety and feasibility of ultrasound-triggered targeted drug delivery of doxorubicin from thermosensitive liposomes in liver tumours (tardox): A single-centre, open-label, phase 1 trial. Lancet Oncol. 2018, 19, 1027–1039.
- Haemmerich, D. Non-invasive image-guided targeted drug delivery. Lancet Oncol. 2018, 19, 1000–1001.
- Borys, N.; Dewhirst, M.W. Drug development of lyso-thermosensitive liposomal doxorubicin: Combining hyperthermia and thermosensitive drug delivery. Adv. Drug Deliv. Rev. 2021, 178, 113985.
- Lindner, L.H.; Eichhorn, M.E.; Eibl, H.; Teichert, N.; Schmitt-Sody, M.; Issels, R.D.; Dellian, M. Novel temperature-sensitive liposomes with prolonged circulation time. Clin. Cancer Res. 2004, 10, 2168–2178.
- Lindner, L.H.; Hossann, M.; Vogeser, M.; Teichert, N.; Wachholz, K.; Eibl, H.; Hiddemann, W.; Issels, R.D. Dual role of hexadecylphosphocholine (miltefosine) in thermosensitive liposomes: Active ingredient and mediator of drug release. J. Control. Release Off. J. Control. Release Soc. 2008, 125, 112–120.
- Lu, T.; Lokerse, W.J.M.; Seynhaeve, A.L.B.; Koning, G.A.; Ten Hagen, T.L.M. Formulation and optimization of idarubicin thermosensitive liposomes provides ultrafast triggered release at mild hyperthermia and improves tumor response. J. Control. Release Off. J. Control. Release Soc. 2015, 220, 425–437.
- Li, L.; ten Hagen, T.L.; Schipper, D.; Wijnberg, T.M.; van Rhoon, G.C.; Eggermont, A.M.; Lindner, L.H.; Koning, G.A. Triggered content release from optimized stealth thermosensitive liposomes using mild hyperthermia. J. Control. Release Off. J. Control. Release Soc. 2010, 143, 274–279.
- Gasselhuber, A.; Dreher, M.R.; Negussie, A.; Wood, B.J.; Rattay, F.; Haemmerich, D. Mathematical spatio-temporal model of drug delivery from low temperature sensitive liposomes during radiofrequency tumour ablation. Int. J. Hyperth. 2010, 26, 499–513.
- Negussie, A.H.; Yarmolenko, P.S.; Partanen, A.; Ranjan, A.; Jacobs, G.; Woods, D.; Bryant, H.; Thomasson, D.; Dewhirst, M.W.; Wood, B.J.; et al. Formulation and characterisation of magnetic resonance imageable thermally sensitive liposomes for use with magnetic resonance-guided high intensity focused ultrasound. Int. J. Hyperth. 2011, 27, 140–155.
- de Smet, M.; Heijman, E.; Langereis, S.; Hijnen, N.M.; Grull, H. Magnetic resonance imaging of high intensity focused ultrasound mediated drug delivery from temperature-sensitive liposomes: An in vivo proof-of-concept study. J. Control. Release Off. J. Control. Release Soc. 2011, 150, 102–110.
- Tucci, S.T.; Kheirolomoom, A.; Ingham, E.S.; Mahakian, L.M.; Tam, S.M.; Foiret, J.; Hubbard, N.E.; Borowsky, A.D.; Baikoghli, M.; Cheng, R.H.; et al. Tumor-specific delivery of gemcitabine with activatable liposomes. J. Control. Release Off. J. Control. Release Soc. 2019, 309, 277–288.
- Asemani, D.; Motamarry, A.; Haemmerich, D. In vitro measurement of release kinetics of temperature sensitive liposomes with a fluorescence imaging system. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2018, 2018, 3216–3219.
- Ranjan, A.; Jacobs, G.C.; Woods, D.L.; Negussie, A.H.; Partanen, A.; Yarmolenko, P.S.; Gacchina, C.E.; Sharma, K.V.; Frenkel, V.; Wood, B.J.; et al. Image-guided drug delivery with magnetic resonance guided high intensity focused ultrasound and temperature sensitive liposomes in a rabbit vx2 tumor model. J. Control. Release 2012, 158, 487–494.
- Staruch, R.M.; Ganguly, M.; Tannock, I.F.; Hynynen, K.; Chopra, R. Enhanced drug delivery in rabbit vx2 tumours using thermosensitive liposomes and mri-controlled focused ultrasound hyperthermia. Int. J. Hyperth. 2012, 28, 776–787.
- Swenson, C.E.; Haemmerich, D.; Maul, D.H.; Knox, B.; Ehrhart, N.; Reed, R.A. Increased duration of heating boosts local drug deposition during radiofrequency ablation in combination with thermally sensitive liposomes (thermodox) in a porcine model. PLoS One 2015, 10, e0139752.
- Motamarry, A.; Wolfe, A.M.; Ramajayam, K.K.; Pattanaik, S.; Benton, T.; Peterson, Y.; Faridi, P.; Prakash, P.; Twombley, K.; Haemmerich, D. Extracorporeal removal of thermosensitive liposomal doxorubicin from systemic circulation after tumor delivery to reduce toxicities. Cancers 2022, 14, 1322.
- Bredlau, A.L.; Motamarry, A.; Chen, C.; McCrackin, M.A.; Helke, K.; Armeson, K.E.; Bynum, K.; Broome, A.M.; Haemmerich, D. Localized delivery of therapeutic doxorubicin dose across the canine blood-brain barrier with hyperthermia and temperature sensitive liposomes. Drug Deliv. 2018, 25, 973–984.
- Park, S.M.; Kim, M.S.; Park, S.J.; Park, E.S.; Choi, K.S.; Kim, Y.S.; Kim, H.R. Novel temperature-triggered liposome with high stability: Formulation, in vitro evaluation, and in vivo study combined with high-intensity focused ultrasound (hifu). J. Control. Release Off. J. Control. Release Soc. 2013, 170, 373–379.
- Hossann, M.; Hirschberger, J.; Schmidt, R.; Baumgartner, C.; Zimmermann, K.; Baer, S.; Ratzlaff, C.; Peller, M.; Troedson, K.; Limmer, S.; et al. A heat-activated drug delivery platform based on phosphatidyl-(oligo)-glycerol nanocarrier for effective cancer treatment. Adv. NanoBiomed Res. 2021, 1, 2000089.
- Hossann, M.; Wiggenhorn, M.; Schwerdt, A.; Wachholz, K.; Teichert, N.; Eibl, H.; Issels, R.D.; Lindner, L.H. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim. Biophys. Acta. 2007, 1768, 2491–2499.
- Hossann, M.; Wang, T.; Wiggenhorn, M.; Schmidt, R.; Zengerle, A.; Winter, G.; Eibl, H.; Peller, M.; Reiser, M.; Issels, R.D.; et al. Size of thermosensitive liposomes influences content release. J. Control. Release Off. J. Control. Release Soc. 2010, 147, 436–443.
- van Valenberg, F.J.P.; Brummelhuis, I.S.G.; Lindner, L.H.; Kuhnle, F.; Wedmann, B.; Schweizer, P.; Hossann, M.; Witjes, J.A.; Oosterwijk, E. Dppg2-based thermosensitive liposomes with encapsulated doxorubicin combined with hyperthermia lead to higher doxorubicin concentrations in the bladder compared to conventional application in pigs: A rationale for the treatment of muscle-invasive bladder cancer. Int. J. Nanomed. 2021, 16, 75–88.
- Zimmermann, K.; Hossann, M.; Hirschberger, J.; Troedson, K.; Peller, M.; Schneider, M.; Bruhschwein, A.; Meyer-Lindenberg, A.; Wess, G.; Wergin, M.; et al. A pilot trial of doxorubicin containing phosphatidyldiglycerol based thermosensitive liposomes in spontaneous feline soft tissue sarcoma. Int. J. Hyperth. 2016, 33, 178–190.
- Kono, K.; Ozawa, T.; Yoshida, T.; Ozaki, F.; Ishizaka, Y.; Maruyama, K.; Kojima, C.; Harada, A.; Aoshima, S. Highly temperature-sensitive liposomes based on a thermosensitive block copolymer for tumor-specific chemotherapy. Biomaterials 2010, 31, 7096–7105.
- Tagami, T.; Ernsting, M.J.; Li, S.D. Efficient tumor regression by a single and low dose treatment with a novel and enhanced formulation of thermosensitive liposomal doxorubicin. J. Control. Release Off. J. Control. Release Soc. 2011, 152, 303–309.
- van Elk, M.; Deckers, R.; Oerlemans, C.; Shi, Y.; Storm, G.; Vermonden, T.; Hennink, W.E. Triggered release of doxorubicin from temperature-sensitive poly(n-(2-hydroxypropyl)-methacrylamide mono/dilactate) grafted liposomes. Biomacromolecules 2014, 15, 1002–1009.
- Iga, K.; Hamaguchi, N.; Igari, Y.; Ogawa, Y.; Toguchi, H.; Shimamoto, T. Increased tumor cisplatin levels in heated tumors in mice after administration of thermosensitive, large unilamellar vesicles encapsulating cisplatin. J. Pharm. Sci. 1991, 80, 522–525.
- Wu, Y.; Yang, Y.; Zhang, F.C.; Wu, C.; Lu, W.L.; Mei, X.G. Epirubicin-encapsulated long-circulating thermosensitive liposome improves pharmacokinetics and antitumor therapeutic efficacy in animals. J. Liposome Res. 2011, 21, 221–228.
- Rossmann, C.; McCrackin, M.A.; Armeson, K.E.; Haemmerich, D. Temperature sensitive liposomes combined with thermal ablation: Effects of duration and timing of heating in mathematical models and in vivo. PLoS One 2017, 12, e0179131.
- Ramajayam, K.K.; Wolfe, A.M.; Motamarry, A.; Nahhas, G.J.; Yost, J.; Yost, M.J.; Haemmerich, D. Untargeted large volume hyperthermia reduces tumor drug uptake from thermosensitive liposomes. IEEE Open J. Eng. Med. Biol. 2021, 2, 187–197.
- Sebeke, L.C.; Castillo Gómez, J.D.; Heijman, E.; Rademann, P.; Simon, A.C.; Ekdawi, S.; Vlachakis, S.; Toker, D.; Mink, B.L.; Schubert-Quecke, C.; et al. Hyperthermia-induced doxorubicin delivery from thermosensitive liposomes via mr-hifu in a pig model. J. Control. Release 2022, 343, 798–812.
- Lokerse, W.J.; Kneepkens, E.C.; Ten Hagen, T.L.; Eggermont, A.M.; Grull, H.; Koning, G.A. In depth study on thermosensitive liposomes: Optimizing formulations for tumor specific therapy and in vitro to in vivo relations. Biomaterials 2016, 82, 138–150.
- Aston, W.J.; Hope, D.E.; Nowak, A.K.; Robinson, B.W.; Lake, R.A.; Lesterhuis, W.J. A systematic investigation of the maximum tolerated dose of cytotoxic chemotherapy with and without supportive care in mice. BMC Cancer 2017, 17, 1–10.
This entry is offline, you can click here to edit this entry!