Herpes simplex virus type 1 (HSV-1) is a neurotropic virus that occasionally may spread to the central nervous system (CNS), being the most common cause of sporadic encephalitis. One of the main neurovirulence factors of HSV-1 is the protein ICP34.5, which although it initially seems to be relevant only in neuronal infections, it can also promote viral replication in non-neuronal cells.
1. Introduction
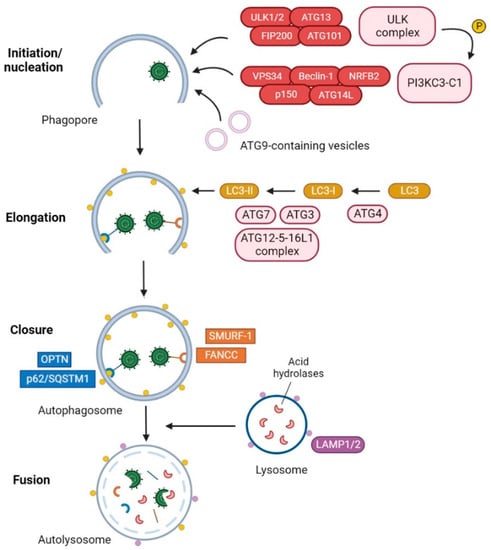
Autophagy [
1] is a highly conserved catabolic process among eukaryotes, consisting of the degradation of intracellular components into lysosomes to ensure metabolic homeostasis [
2,
3]. There are different types of autophagic pathways: macroautophagy, microautophagy, endosomal microautophagy and chaperone-mediated autophagy (CMA) [
4]. The most studied pathway is macroautophagy, which involves the de novo generation of double-membrane vesicles called autophagosomes, which are subsequently fused with lysosomes for cargo degradation. Autophagosome formation is a highly regulated process carried out by functional units constituted by autophagy-related (ATG) proteins [
5]. This process consists of five sequential steps, which include initiation, nucleation, elongation, closure, and fusion (
Figure 1).
Figure 1. Degradation of HSV-1 by selective autophagy. The ULK complex initiates the formation of the phagophore by phosphorylating components of the PI3KC3 complex I and mediating the trafficking of ATG9-containing vesicles. The LC3-PE conjugation system participates in the elongation and closure of the double membrane, resulting in the formation of the autophagosome. The selective autophagy receptors (SARs) p62/SQSTM1 [
21] and optineurin (OPTN) [
22] and the autophagy receptor-like factors Fanconi anemia group C protein (FANCC) [
23] and SMAD ubiquitin regulatory factor 1 (SMURF-1) [
21] can interact with HSV-1 and mediate the recruitment of HSV-1 virions and/or viral cytoplasmic components into the autophagosomes. Once cargo has been engulfed, the external membrane of the autophagosome fuses with a lysosome for degradation of viral components.
Autophagy can be classified into non-selective or selective types depending on the cargo. In non-selective autophagy, bulk portions of the cytoplasm are sequestered by phagophores. This type of autophagy is usually stimulated under starvation conditions, due to its contribution to the maintenance of nutrient levels in cells. On the other hand, selective autophagy is based on the degradation of specific intracellular cargo through the activity of selective autophagy receptors (SARs). SARs interact with ATG8 family proteins on the inner membrane surface of phagophores, mediating the delivery of specific cargo and facilitating the recruitment of autophagic machinery [
24,
25].
Many viruses can reverse and exploit multiple steps of autophagy, thus evading the immune response and facilitating viral replication [
29]. This is the case of herpes simplex virus type 1 (HSV-1), which has acquired the ability to down-regulate the autophagic pathway. HSV-1 is a neurotropic virus that, after a primary infection in epithelial cells, traffics from axon terminals to the trigeminal ganglia and establishes latency [
30].
2. The HSV-1 Factor of Virulence ICP34.5
2.1. The HSV-1 γ34.5 Gene and the Infected Cell Protein 34.5
The infected-cell protein 34.5 (ICP34.5) of HSV-1 is encoded by the γ34.5 gene, also known as RL1. This gene is located in the terminal repeat (TR) sequences of the HSV-1 DNA, being present in two copies per genome [
36]. The γ34.5 gene is classified as a gamma-1 or leaky late gene. Although maximal levels of ICP34.5 are reached in the late phase of infection, the expression and function of the protein at early times, 3 hours post-infection, are essential for the control of the host cell environment and for successful viral replication [
37]. The γ34.5 gene lacks a canonical TATAA box. Part of its promoter and the transcription initiation site are within the unique repeat junction called the “a” sequence, and its 5′unstranscribed domain (UTR 5′) is GC rich and has several repeats, lacking the characteristic features of HSV-1 promoters [
38].
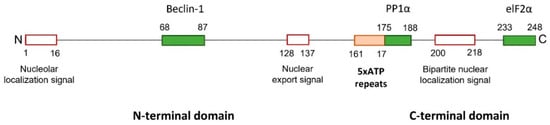
The structure of ICP34.5 consists of a disordered N-terminal domain, followed by a linker region with a variable number of ATP (Ala-Thr-Pro) repeats, and a conserved C-terminal domain (Figure 2).
Figure 2. ICP34.5 of HSV-1 strain 17. HSV-1 ICP34.5 is a protein with 248 amino acids that can be divided into three regions: the amino and the carboxyl terminal domains, and a linked tandem of five ATP repeats (161–175 aa) (orange box). The N-terminal region of ICP34.5 contains a nucleolar localization and nuclear export signals, and the C-terminal domain includes a nuclear localization signal (red boxes). Three binding domains (green boxes) have been characterized: the Beclin-1-binding domain (BBD) in the N-terminus, and the PP1α- and elF2α-binding domains in the C-terminus.
ICP34.5 is a highly dynamic protein that shuttles between the nucleus, the nucleolus, and the cytoplasm, with its function dependent on cellular location [
42]. Generally, during early times of HSV-1 infection, the protein is mainly located in the nucleus, and, at 8–12 h post-infection, it is accumulated in the cytoplasm [
43]. The N-terminus of ICP34.5 contains an arginine-rich cluster, which primarily targets the protein of the nucleolus, and a leucine-rich cluster, which acts as a nuclear export signal. The C-terminus contains a bipartite nuclear localization signal, consisting of two clusters of basic amino acids separated by a 9–11 amino acids spacer, which is implied in the import of ICP34.5 into the nucleus [
42].
2.2. HSV-1 ICP34.5: A Catch-All
ICP34.5 is primarily described as a neurovirulence factor essential for HSV-1 infection of neurons in vivo. HSV-1 lacking the γ34.5 gene is profoundly neuroattenuated in mouse brains [
36,
46]. However, the role of ICP34.5 in viral pathogenesis is strongly dependent on both cell type and cell stage, and it is not only restricted to neuronal cells. For instance, ICP34.5 is required for the HSV-1 infection of stationary-state mouse embryo fibroblast 3T6 cells, whereas the replication of ICP34.5-deficient HSV-1 in hamster kidney fibroblasts BHK cells is indistinguishable from the wild-type virus [
47]. The role of ICP34.5 in HSV-1 infection is extremely complex, and the reason for the strong cell type dependence is largely unknown.
2.2.1. Functions of the Carboxyl Domain of HSV-1 ICP34.5
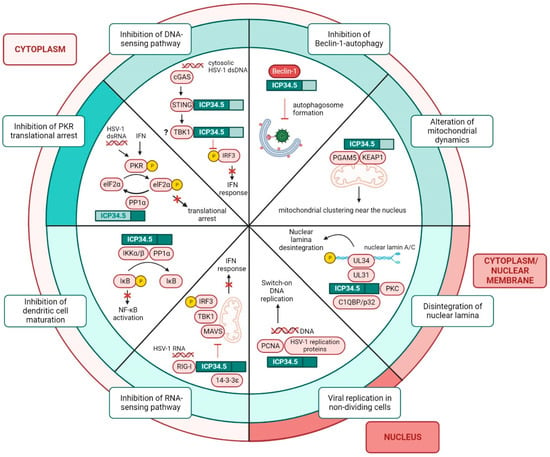
The first function of ICP34.5 to be described is associated with the C-terminal domain. Upon viral infection, host cells detect the presence of the virus through pattern recognition receptors (PRRs). PRRs recognize conserved viral structures called pathogen-associated molecular patterns (PAMPs), which include viral proteins, DNA and RNA [
49]. PRRs relay signals to TANK-binding kinase 1 (TBK1), which phosphorylates the interferon (IFN) regulatory factor 3/7 (IRF3/7). Phosphorylated IRF3/7 is translocated to the nucleus to induce the production of type I IFN and other cytokines. In response to IFN, several molecules are activated through the JAK/STAT pathway, establishing an antiviral environment in the neighboring cells [
50,
51]. Among these antiviral molecules is dsRNA-dependent protein kinase (PKR), a cytoplasmic protein capable of detecting viral dsRNA. Its major role is the inhibition of both cellular and viral protein synthesis through the phosphorylation of the translation initiation factor 2 subunit α (elF2α) [
52]. HSV-1 can block this important antiviral mechanism through ICP34.5 [
53] (
Figure 3).
Figure 3. Functions of ICP34.5 in HSV-1 infected cells. Functions of ICP34.5 can be classified according to the cellular location of the protein, which shuttles between the cytoplasm and the nucleus. The amino and the carboxyl domains of ICP34.5 play different roles in infected cells. The C-terminus prevents the translational arrest by the binding of PP1α and the subsequent dephosphorylation of elF2α. The N-terminus is involved in the suppression of Beclin-1-autophagy, the degradation of the nuclear lamina to facilitate nucleocapsid egress from the nucleus, and the blockade of IFN response by the inhibition of the dsDNA-sensing pathway. The full-length protein plays a role in virus replication in non-dividing cells, in the prevention of DCs maturation, and in the suppression of the IFN response by blocking the RNA-sensing pathway.
2.2.2. Functions of the Amino Domain of HSV-1 ICP34.5
The functions of ICP34.5 associated with its N-terminal domain (
Figure 3) remain obscure. On the one hand, the 68–87 amino acid region of ICP34.5 or Beclin-1-binding domain (BBD) recruits and blocks an essential protein for autophagosome biogenesis, Beclin-1, thus inhibiting autophagy [
57]. Initially, the function of BBD was understood to be restricted to the prevention of autophagy, but an additional role of this domain was recently discovered. BBD not only interacts with Beclin-1, but also with multiple regulators of the antioxidant response, mitochondrial trafficking, and programmed cell death. In HSV-1-infected primary mouse neurons, the interaction of BBD with the sensor of oxidative stress KEAP1 and the regulator of mitochondrial dynamics PGAM5 causes the formation of mitochondrial clusters near the nucleus, where virions are produced. This mitochondrial distribution relieves the bioenergetic demands on the cell during infection and facilitates transcription under oxidative stress [
58].
On the other hand, the N-terminal domain of ICP34.5 plays an important role in the fight against IFN responses by the dephosphorylation of IRF3. Initially, it was described that TBK1 interacts with ICP34.5, thereby inhibiting IRF3 phosphorylation. ICP34.5 was observed to bind to TBK1 in the 76–106 amino acid region of the N-terminus, known as the TBK-1-binding domain (TBD). HSV-1 lacking the γ34.5 gene replicates efficiently in TBK1(-/-) cells but not in TBK1(+/+) cells [
59,
60]. However, later studies demonstrated that TBD binds overexpressed but not endogenous TBK1.
2.2.3. Functions of the Amino and Carboxyl Domains of HSV-1 ICP34.5
Both amino and carboxyl terminal regions are required for certain roles of ICP34.5 (
Figure 3). Among these functions is the suppression of dendritic cells (DCs) maturation. Unlike the wild-type virus, HSV-1 lacking the γ34.5 gene stimulates the expression of MHC-II, CD86 and cytokines in immature DCs [
68]. The mechanism by which ICP34.5 disrupts the maturation of DCs is mediated by the dephosphorylation of IκB kinase. The N-terminal domain of ICP34.5 binds to the protein IKKα/β and the C-terminal domain recruits PP1α. This multiprotein complex dephosphorylates IκB kinase to prevent the activation of NF-κB, a transcription factor that regulates the expression of genes involved in immune response, inflammation, and cell survival [
69].
3. HSV-1 Modulation of Autophagy
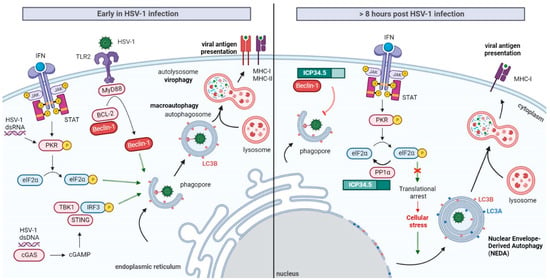
Induction of Autophagy at the Early Stages of HSV-1 Infection
Under stress conditions, such as nutrient starvation, oxidative stress, the presence of unfolded proteins or pathogen infections, autophagy can be stimulated to promote metabolic adaptation and cellular survival. As a result of the role of this pathway in cellular protection against pathogens, it is not surprising that, in early stages of HSV-1 infection, autophagy flux is usually stimulated (Figure 4). However, the molecular mechanisms involved in autophagy stimulation during HSV-1 infection are still not fully understood. The apparent cell-type dependence of these mechanisms makes their research strongly complicated.
Figure 4. Autophagy modulation by ICP34.5 in HSV-1 infection. Early in HSV-1 infection, detection of the virus by host cell promotes the stimulation of macroautophagy. This pathway may act as a cellular defense mechanism involved in virophagy and the processing of viral antigen for MHC presentation. Autophagic flux can be induced in manner that is dependent on viral gene expression by the PKR/elF2α pathway. Autophagy may be enhanced through the recognition of PAMPs by the surface receptor TLR2. Activated TLR2 recruits the adaptor MyD88, which causes the dissociation of Beclin-1 from the BCL-2 inhibitory complex, resulting in the induction of autophagy. Finally, HSV-1 dsDNA may be recognized by the cytosolic dsDNA-sensing cGAS, which produces cGAMP to activate STING and TBK1. Activation of TBK1 promotes autophagy stimulation. To deal with virophagy, HSV-1 suppresses macroautophagy by blocking Beclin-1 through ICP34.5. Besides, HSV-1 prevents the PKR translational arrest by the recruitment of elF2 and PP1 in the C-terminus of ICP34.5. As a general stress response, the host cell initiates an alternative autophagy pathway known as NEDA. This process consists of the Beclin-1-independent formation of four-layered membrane structures by the coiling of the nuclear membrane. NEDA is suggested to have an antiviral role promoting viral antigen presentation, but further research about the cellular functions of NEDA is required.
In murine embryonic fibroblasts and sympathetic neurons, the IFN-inducible PKR signaling pathway not only leads to cellular translational arrest, but also to the induction of autophagy and HSV-1 degradation into autolysosomes. The stimulation of autophagy by this pathway appears to be dependent on HSV-1 gene expression [
71,
72,
73]. Viral gene expression is also necessary for the induction of autophagy in monocytic-derived DCs, because infection with UV-inactivated HSV-1 does not lead to the stimulation of this degradative pathway [
74]. The relevance of PKR mediating selective autophagy has been demonstrated not only in vitro, but also in vivo [
57].
4. Selective Inhibition of Autophagy by HSV-1 ICP34.5
Upon the initial induction of autophagy, HSV-1 may counteract host cells by down-modulating the pathway through ICP34.5. The role of ICP34.5 in autophagy inhibition is restricted to a specific domain (amino acids 68 to 87) implicated in the recruitment of Beclin-1 [
57], an essential protein for autophagosome formation [
85]. This domain is known as “Beclin-1-binding domain” or BBD. The C-terminal region of the protein is dispensable for this autophagy-inhibitory activity, suggesting that the modulation of autophagy is independent of PP1α/elF2α binding to ICP34.5 [
57].
The effect of HSV-1 ICP34.5 on autophagy is strongly dependent on the cell type. Although BBD inhibits the formation of autophagosomes in neurons and fibroblasts [
73], in bone-marrow DCs (BM-DCs) it prevents autophagosome maturation, causing the accumulation of long-lived proteins and autophagosomes [
86]. In some cell types, including corneal epithelial and retinal ganglion cells, the protein has no effect on cellular autophagy [
87].
Autophagy suppression by the virus not only prevents virion removal into autolysosomes, but it also prevents the processing and delivery of intracellular viral antigens to MHC class I and II molecules, inhibiting adaptative immunity [
88] (
Figure 4). When murine DCs are infected with ICP34.5-deficient HSV-1, viral antigen presentation on MHC-I is increased compared to the wild-type virus [
89]. In mice, HSV-1 lacking BBD precludes autophagy-mediated MHC-II antigen presentation, decreasing the stimulation of CD4+ T cells [
90].
The reduced presentation of viral antigens, a consequence of HSV-1 autophagy arrest, may be compensated by host cells by an alternative Beclin-1-independent autophagic pathway. Recently, the presence of four-layered membrane structures, containing the autophagy marker LC3B, was described in HSV-1 infected macrophages. These four-layered structures are formed by rolling up the inner and outer nuclear membrane, and they are accumulated in the cytoplasm about eight hours post-infection. These vacuole structures enhance the presentation of endogenous viral antigens on MHC-I molecules, providing an additional pathway apart from viral antigen degradation by the proteasome. Whereas the formation of canonical autophagosomes can be promoted by pharmacological autophagy inducers, the generation of these four-layered structures is only induced by HSV-1 infection, which means that it is regulated in a way that is different from macroautophagy [
88]. This autophagic pathway was denominated as NEDA (Nuclear-Envelope Derived Autophagy) (
Figure 4).
5. The Intricate Role of the Beclin-1-Binding Domain of ICP34.5 in HSV-1 Virulence
HSV-1 strains lacking the BBD of ICP34.5 exhibit reduced neurovirulence and viral replication in mice compared to wild-type viruses [
57,
61,
90]. However, whereas HSV-1 lacking BBD is strongly neuroattenuated in the brains of mice, this protein domain is dispensable for productive HSV-1 replication in the neuronal established cell line SK-N-SH [
57]. Even in primary fibroblast cultures, no difference is noticeable in viral replication between HSV-1 lacking BBD and the wild-type virus [
92].
HSV-1 lacking BBD is not as attenuated in vitro as the data would predict. Recently, it has been proposed that the differences in virulence between in vivo and in vitro studies could be due to the presence of ferric nitrate in culture media. In the presence of this iron salt, which is involved in redox reactions, the attenuated replication of HSV-1 lacking BBD in primary human fibroblasts is partially restored. The cause of the masked BBD effect can be explained by the fact that this domain recruits not only Beclin-1, but also multiple mitochondrial regulators that play a relevant role in redox reactions and cellular metabolism [
58]. Unfortunately, primary neuron cultures do not survive in redox buffer-free medium conditions. Hence, viral replication experiments cannot be performed under these conditions, where the effect of BBD is not masked.
6. ICP34.5 Is Not Alone in HSV-1 Autophagy Inhibition
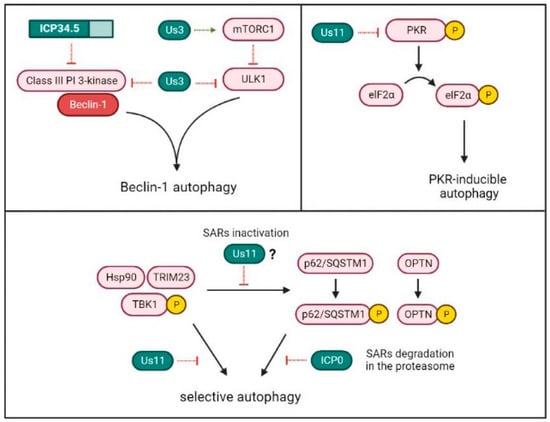
HSV-1 exploits the factor of virulence ICP34.5 to inhibit Beclin-1-dependent autophagy. However, ICP34.5 is neither the unique HSV-1 protein capable of modulating autophagy, nor is Beclin-1 the only target used by the virus for autophagy suppression (Figure 5).
Figure 5. HSV-1 mechanisms for autophagy inhibition. HSV-1 proteins Us3 and ICP34.5 can inhibit macroautophagy by binding to Beclin-1, which is required for autophagosome formation. Us3 can also suppress the pathway by the activation of the negative regulator of autophagy mTORC1 or by the inactivation of the ULK1 complex, which is involved in phagophore initiation. The HSV-1 protein Us11 can prevent the induction of autophagy mediated by the PKR/elF2α pathway. Besides, Us11 inhibits virus-induced autophagy by the disassembly of the TRIM23-Hsp90-TBK1 complex. Finally, the HSV-1 protein ICP0 promotes the degradation in the proteasome of the selective autophagy receptors p62/SQSTM1 and OPTN, which are implied in the recruitment of HSV-1 in autophagosomal membranes. The TRIM23-TBK1 complex is involved in the phosphorylation and activation of these selective receptors. However, the inhibitory role of Us11 on the TRIM23-TBK1 complex has not been directly associated with a possible SARs inactivation.
Us11 is an HSV-1 late protein that binds to dsRNA and physically interacts with PKR [
93]. Through the direct association to PKR, Us11 prevents the activation of the PKR/elF2α signaling pathway and, consequently, translational arrest [
94,
95]. Although Us11 is not able to interact with Beclin-1, its interaction with PKR has a strong anti-autophagic activity with cells. Thus, autophagy inhibition in HSV-1 infected cells can be a result of the activity not only from ICP34.5, but also from Us11 [
96].
In addition to preventing autophagosome formation, recent research suggests that viruses can evade virophagy by targeting SARs [
100]. HSV-1 may down-modulate two important SARs implied in the recognition and delivery of specific viral ubiquitinated cargo to the phagophores: the autophagy receptor sequestosome 1 (p62/SQSTM1) and the mitophagy adaptor optineurin (OPTN). Cellular levels of p62/SQSTM1 and OPTN are significantly reduced after 3–6 h of HSV-1 infection in various cell lines [
101]. After activation by TRIM23, TBK1 phosphorylates and activates p62/SQSTM1 and OPTN [
102].
Recently, it was discovered that the HSV-1 protein Ser/Thr kinase Us3 can also suppress autophagy [105]. Us3 may phosphorylate and activate the nutrient-sensing mammalian target of rapamycin kinase complex 1 (mTORC1) [106], which is a negative regulator of the ULK complex that keeps autophagy inactive under physiological conditions [107]. In addition, Us3 can modulate autophagy downstream of mTORC1, by the phosphorylation and inactivation of both the ULK1 complex and Beclin-1 [105].
7. The Two Sides of Autophagy
Autophagy is often considered a double-edged sword, and a possible proviral role of this pathway in HSV-1 infection was previously raised [115]. The suppression of autophagy, both by pharmacological inhibitors and by siRNA knockdown technology, significantly impairs HSV-1 infection in human acute monocytic leukemia (THP-1) cells and in primary human monocytes. These results indicate that the virus may use the autophagic machinery for its own benefit [80]. One proposed proviral function of autophagy is the degradation of the nuclear lamina [74]. The autophagy protein LC3 is present not only in the cytoplasm, but also in the nucleus, where it can interact directly with the lamins. This interaction can lead to the transport of the nuclear lamins from the nucleus to the cytoplasm for lysosomal degradation [116]. Lamina disintegration by nuclear autophagy has been proposed to facilitate the egress of HSV-1 capsids from the nucleus. This kind of autophagy is not down-modulated by ICP34.5, which means that, similar to NEDA, it is Beclin-1 independent [74].
8. Herpes Simplex Virus Type 2 and Autophagy
Herpes simplex virus type 2 (HSV-2) is an alphaherpesvirus that primarily infects genital epithelial cells and establishes latency in sensory neurons of the sacral dorsal root ganglia. HSV-2 infection can cause severe genital diseases, with this virus being the most common cause of genital ulcers [
124]. Although the modulation of autophagy by HSV-1 has been extensively analyzed, research focused on HSV-2 is scarce. It has been observed that HSV-2 inhibits autophagy in genital epithelial cells and the treatment with an autophagy inducer, known as JZ-1, significantly impairs viral infection in vitro [
125] and in vivo [
126]. Similar to HSV-1, the modulation of autophagy by HSV-2 seems to be cell-type dependent [
47], and basal autophagy appears to play an important role for HSV-2 productive infection [
127].
However, although HSV-2 can inhibit the autophagic flux similarly to HSV-1, the anti-autophagic proteins that are involved in preventing autophagy have not yet been determined. ICP34.5 is an important virulence factor for HSV-2, since deletion of the γ34.5 gene leads to a severe reduction in infection [
128,
129], but it remains unknown if HSV-2 down-modulates autophagy through ICP34.5. The amino acid sequence of the N-terminus of HSV-1 ICP34.5 is only 41% identical to the first exon of HSV-2 ICP34.5, and insertions appear to disrupt the corresponding HSV-1 BBD [
129].
9. Conclusions
Autophagy is a fine-tuned and regulated process, and, when “autophagic balance” tips to one side, HSV-1 infection is often impaired. As a result of the strong influence of this pathway in viral infections, the modulation of autophagy has been suggested as a potential future therapy against HSV-1. However, before becoming fully involved in this type of treatments, it should be considered that unbalanced autophagy can negatively affect not only viral infection, but also cellular homeostasis and survival.
Autophagy is especially important in maintaining the correct functionality of the CNS. Deregulation of this pathway, whether excessive or insufficient, has been related to severe neurodegenerative disorders. Moreover, the suppression of autophagy by HSV-1 in the CNS has been proposed as a cause of neurodegeneration. The “autophagic balance” is extremely sensitive, and further research is required to decode the mechanisms that allow us to modulate autophagy accurately.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232113643