Pancreatic ductal adenocarcinoma (PDAC) is a lethal cancer type as it is prone to metastases and is difficult to diagnose at an early stage. Despite advances in molecular detection, its clinical prognosis remains poor and it is expected to become the second leading cause of cancer-related deaths. Approximately 85% of patients develop glucose metabolism disorders, most commonly diabetes mellitus, within three years prior to their pancreatic cancer diagnosis. Diabetes, or glucose metabolism disorders related to PDAC, are typically associated with insulin resistance, and beta cell damage, among other factors. From the perspective of molecular regulatory mechanisms, glucose metabolism disorders are closely related to PDAC initiation and development and to late invasion and metastasis.
- pancreatci ductal adenocarcinoma
- glycolytci phenotype
- glucose
1. Clinical Association between Pancreatic Ductal Adenocarcinoma and Diabetes
2. Promotion of Pancreatic Ductal Adenocarcinoma through a High Glycolytic Phenotype
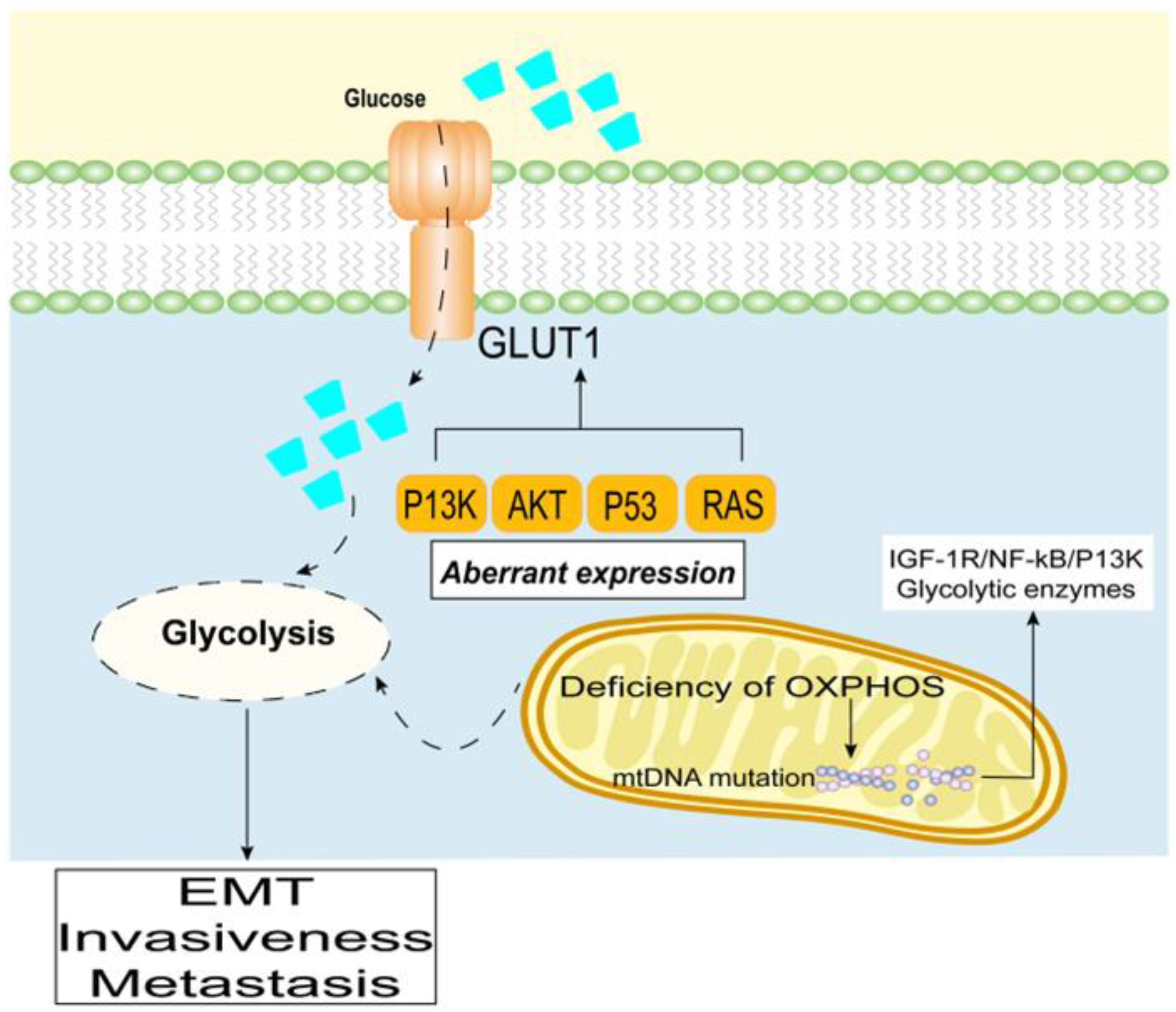
3. Therapeutic Strategies Targeting Glycolysis
| Therapeutic Agent | Therapeutic Strategy | Cancer type | Reference |
|---|---|---|---|
| Prevent risk factors | Weight loss and the adoption of a healthy diet, surgery | Most of malignant tumors | [43][44] |
| Metformin, sulfonylureas, thiazolidinediones (pioglitazone) | Control the blood glucose | Breast, Pancreatic, Colorectal, and Prostate cancers | [45][46] |
| Cardamonin | Inhibit HIF-1α, inhibition of the glycolytic process | Breast cancer | [47] |
| Trametinib, GDC-0623 | Inhibit the MEK pathway | PDAC | [48] |
| Vitamin D | Monitoring and supplementation of 25-hydroxyvitamin D levels is beneficial for treatment | Pancreatic cancer | [49] |
This entry is adapted from the peer-reviewed paper 10.3390/cancers15020498
References
- Gartner, S.; Kruger, J.; Aghdassi, A.A.; Steveling, A.; Simon, P.; Lerch, M.M.; Mayerle, J. Nutrition in Pancreatic Cancer: A Review. Gastrointest. Tumors 2016, 2, 195–202.
- Pannala, R.; Leirness, J.B.; Bamlet, W.R.; Basu, A.; Petersen, G.M.; Chari, S.T. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008, 134, 981–987.
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861.
- Inoue, M.; Iwasaki, M.; Otani, T.; Sasazuki, S.; Noda, M.; Tsugane, S. Diabetes mellitus and the risk of cancer: Results from a large-scale population-based cohort study in Japan. Arch. Intern. Med. 2006, 166, 1871–1877.
- Saydah, S.H.; Platz, E.A.; Rifai, N.; Pollak, M.N.; Brancati, F.L.; Helzlsouer, K.J. Association of markers of insulin and glucose control with subsequent colorectal cancer risk. Cancer Epidemiol. Biomark. Prev. 2003, 12, 412–418.
- Stolzenberg-Solomon, R.Z.; Graubard, B.I.; Chari, S.; Limburg, P.; Taylor, P.R.; Virtamo, J.; Albanes, D. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA 2005, 294, 2872–2878.
- Mizuno, S.; Nakai, Y.; Isayama, H.; Takahara, N.; Miyabayashi, K.; Yamamoto, K.; Kawakubo, K.; Mohri, D.; Kogure, H.; Sasaki, T.; et al. Diabetes is a useful diagnostic clue to improve the prognosis of pancreatic cancer. Pancreatology 2013, 13, 285–289.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. S1), S67–S74.
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342.
- Korc, M. Pancreatic cancer-associated diabetes is an “exosomopathy”. Clin. Cancer Res. 2015, 21, 1508–1510.
- Daryabor, G.; Atashzar, M.R.; Kabelitz, D.; Meri, S.; Kalantar, K. The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front. Immunol. 2020, 11, 1582.
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37.
- Eibl, G.; Cruz-Monserrate, Z.; Korc, M.; Petrov, M.S.; Goodarzi, M.O.; Fisher, W.E.; Habtezion, A.; Lugea, A.; Pandol, S.J.; Hart, P.A.; et al. Diabetes Mellitus and Obesity as Risk Factors for Pancreatic Cancer. J. Acad. Nutr. Diet. 2018, 118, 555–567.
- Hart, P.A.; Bellin, M.D.; Andersen, D.K.; Bradley, D.; Cruz-Monserrate, Z.; Forsmark, C.E.; Goodarzi, M.O.; Habtezion, A.; Korc, M.; Kudva, Y.C.; et al. Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol. Hepatol. 2016, 1, 226–237.
- Sah, R.P.; Nagpal, S.J.; Mukhopadhyay, D.; Chari, S.T. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 423–433.
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129.
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12.
- Liu, Y.H.; Hu, C.M.; Hsu, Y.S.; Lee, W.H. Interplays of glucose metabolism and KRAS mutation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 817.
- Chikamoto, A.; Inoue, R.; Komohara, Y.; Sakamaki, K.; Hashimoto, D.; Shiraishi, S.; Takamori, H.; Yamashita, Y.I.; Yoshida, N.; Yamanaka, T.; et al. Preoperative High Maximum Standardized Uptake Value in Association with Glucose Transporter 1 Predicts Poor Prognosis in Pancreatic Cancer. Ann. Surg. Oncol. 2017, 24, 2040–2046.
- Fujita, Y.; Matsuda, S.; Sasaki, Y.; Masugi, Y.; Kitago, M.; Yagi, H.; Abe, Y.; Shinoda, M.; Tokino, T.; Sakamoto, M.; et al. Pathogenesis of multiple pancreatic cancers involves multicentric carcinogenesis and intrapancreatic metastasis. Cancer Sci. 2020, 111, 739–748.
- Liu, Z.; Hayashi, H.; Matsumura, K.; Ogata, Y.; Sato, H.; Shiraishi, Y.; Uemura, N.; Miyata, T.; Higashi, T.; Nakagawa, S.; et al. Hyperglycaemia induces metabolic reprogramming into a glycolytic phenotype and promotes epithelial-mesenchymal transitions via YAP/TAZ-Hedgehog signalling axis in pancreatic cancer. Br. J. Cancer 2022.
- Li, M.; Bu, X.; Cai, B.; Liang, P.; Li, K.; Qu, X.; Shen, L. Biological role of metabolic reprogramming of cancer cells during epithelial-mesenchymal transition (Review). Oncol. Rep. 2019, 41, 727–741.
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial(-)Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042.
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50.
- Espiau-Romera, P.; Courtois, S.; Parejo-Alonso, B.; Sancho, P. Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification. J. Clin. Med. 2020, 9, 4128.
- Hardie, R.A.; van Dam, E.; Cowley, M.; Han, T.L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; McKenna, J.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 2.
- Carmona-Carmona, C.A.; Dalla Pozza, E.; Ambrosini, G.; Errico, A.; Dando, I. Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 4128.
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19.
- Song, W.; He, X.; Gong, P.; Yang, Y.; Huang, S.; Zeng, Y.; Wei, L.; Zhang, J. Glycolysis-Related Gene Expression Profiling Screen for Prognostic Risk Signature of Pancreatic Ductal Adenocarcinoma. Front. Genet. 2021, 12, 639246.
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21.
- Guo, Q. Changes in mitochondrial function during EMT induced by TGFbeta-1 in pancreatic cancer. Oncol. Lett. 2017, 13, 1575–1580.
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591.
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32.
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392.
- Zaidieh, T.; Smith, J.R.; Ball, K.E.; An, Q. Mitochondrial DNA abnormalities provide mechanistic insight and predict reactive oxygen species-stimulating drug efficacy. BMC Cancer 2021, 21, 427.
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal 2020, 33, 435–454.
- Krapf, S.A.; Lund, J.; Lundkvist, M.; Dale, M.G.; Nyman, T.A.; Thoresen, G.H.; Kase, E.T. Pancreatic cancer cells show lower oleic acid oxidation and their conditioned medium inhibits oleic acid oxidation in human myotubes. Pancreatology 2020, 20, 676–682.
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435.
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812.
- Schauer, D.P.; Feigelson, H.S.; Koebnick, C.; Caan, B.; Weinmann, S.; Leonard, A.C.; Powers, J.D.; Yenumula, P.R.; Arterburn, D.E. Bariatric Surgery and the Risk of Cancer in a Large Multisite Cohort. Ann. Surg. 2019, 269, 95–101.
- George, S.; Jean-Baptiste, W.; Yusuf Ali, A.; Inyang, B.; Koshy, F.S.; George, K.; Poudel, P.; Chalasani, R.; Goonathilake, M.R.; Waqar, S.; et al. The Role of Type 2 Diabetes in Pancreatic Cancer. Cureus 2022, 14, e26288.
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pr. 2018, 143, 409–419.
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin inhibits breast cancer growth by repressing HIF-1alpha-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 377.
- Yan, L.; Tu, B.; Yao, J.; Gong, J.; Carugo, A.; Bristow, C.A.; Wang, Q.; Zhu, C.; Dai, B.; Kang, Y.; et al. Targeting Glucose Metabolism Sensitizes Pancreatic Cancer to MEK Inhibition. Cancer Res. 2021, 81, 4054–4065.
- Klapdor, S.; Richter, E.; Klapdor, R. Vitamin D status and per-oral vitamin D supplementation in patients suffering from chronic pancreatitis and pancreatic cancer disease. Anticancer. Res. 2012, 32, 1991–1998.
- Ewald, N.; Bretzel, R.G. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur. J. Intern. Med. 2013, 24, 203–206.