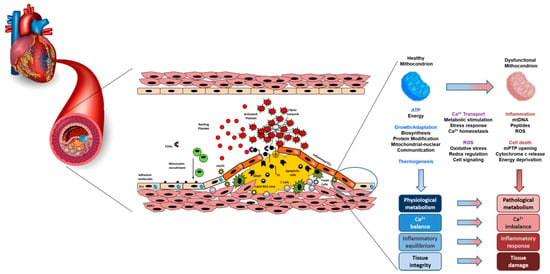
Atherosclerosis is a multifactorial inflammatory pathology that involves metabolic processes. Improvements in therapy have drastically reduced the prognosis of cardiovascular disease. Nevertheless, a significant residual risk is still relevant, and is related to unmet therapeutic targets. Endothelial dysfunction and lipid infiltration are the primary causes of atherosclerotic plaque progression. In this contest, mitochondrial dysfunction can affect arterial wall cells, in particular macrophages, smooth muscle cells, lymphocytes, and endothelial cells, causing an increase in reactive oxygen species (ROS), leading to oxidative stress, chronic inflammation, and intracellular lipid deposition.
- mitochondrial disorders
- atherosclerosis
- endothelium
- cardiovascular disease
1. Introduction
2. Mechanism of Mitochondrial Dysfunction and Molecular Implication for the Cardiovascular System
3. Mitochondrial Dysfunction and Endothelial Dysfunction
This entry is adapted from the peer-reviewed paper 10.3390/ijms24021086
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596.
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021.
- Sanchez-Vinas, A.; Corral-Partearroyo, C.; Gil-Girbau, M.; Penarrubia-Maria, M.T.; Gallardo-Gonzalez, C.; Olmos-Palenzuela, M.D.; Aznar-Lou, I.; Serrano-Blanco, A.; Rubio-Valera, M. Effectiveness and cost-effectiveness of an intervention to improve Initial Medication Adherence to treatments for cardiovascular diseases and diabetes in primary care: Study protocol for a pragmatic cluster randomised controlled trial and economic model (the IMA-cRCT study). BMC Prim. Care 2022, 23, 170.
- Alizadeh, G.; Gholipour, K.; Azami-Aghdash, S.; Dehnavieh, R.; JafarAbadi, M.A.; Azmin, M.; Khodayari-Zarnaq, R. Social, Economic, Technological, and Environmental Factors Affecting Cardiovascular Diseases: A Systematic Review and Thematic Analysis. Int. J. Prev. Med. 2022, 13, 78.
- Lee, C.B.; Liao, C.M.; Peng, L.H.; Lin, C.M. Economic fluctuations and cardiovascular diseases: A multiple-input time series analysis. PLoS ONE 2019, 14, e0219358.
- Aminde, L.N.; Veerman, L. Interventions for the prevention of cardiovascular diseases: A protocol for a systematic review of economic evaluations in low-income and middle-income countries. BMJ Open 2016, 6, e013668.
- Leal, J.; Luengo-Fernandez, R.; Gray, A.; Petersen, S.; Rayner, M. Economic burden of cardiovascular diseases in the enlarged European Union. Eur. Heart J. 2006, 27, 1610–1619.
- Frak, W.; Wojtasinska, A.; Lisinska, W.; Mlynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines 2022, 10, 1938.
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533.
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330.
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxidative Med. Cell. Longev. 2020, 2020, 5245308.
- Greenberg, S.R. Atherosclerosis and mitochondria. Ann. Intern. Med. 1970, 73, 861–862.
- Salnikova, D.; Orekhova, V.; Grechko, A.; Starodubova, A.; Bezsonov, E.; Popkova, T.; Orekhov, A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8990.
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965.
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12.
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. From Mitochondria to Atherosclerosis: The Inflammation Path. Biomedicines 2021, 9, 258.
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137.
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108.
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology 2019, 8, 48.
- Shemiakova, T.; Ivanova, E.; Wu, W.K.; Kirichenko, T.V.; Starodubova, A.V.; Orekhov, A.N. Atherosclerosis as Mitochondriopathy: Repositioning the Disease to Help Finding New Therapies. Front. Cardiovasc. Med. 2021, 8, 660473.
- Jennings, R.B.; Ganote, C.E. Mitochondrial structure and function in acute myocardial ischemic injury. Circ. Res. 1976, 38, I80–I91.
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076.
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726.
- Iglewski, M.; Hill, J.A.; Lavandero, S.; Rothermel, B.A. Mitochondrial fission and autophagy in the normal and diseased heart. Curr. Hypertens. Rep. 2010, 12, 418–425.
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894.
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97.
- Chehaitly, A.; Guihot, A.L.; Proux, C.; Grimaud, L.; Aurriere, J.; Legouriellec, B.; Rivron, J.; Vessieres, E.; Tetaud, C.; Zorzano, A.; et al. Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants 2022, 11, 1078.
- Hubens, W.H.G.; Vallbona-Garcia, A.; de Coo, I.F.M.; van Tienen, F.H.J.; Webers, C.A.B.; Smeets, H.J.M.; Gorgels, T. Blood biomarkers for assessment of mitochondrial dysfunction: An expert review. Mitochondrion 2022, 62, 187–204.
- Xin, Y.; Zhang, X.; Li, J.; Gao, H.; Li, J.; Li, J.; Hu, W.; Li, H. New Insights Into the Role of Mitochondria Quality Control in Ischemic Heart Disease. Front. Cardiovasc. Med. 2021, 8, 774619.
- Garbincius, J.F.; Luongo, T.S.; Jadiya, P.; Hildebrand, A.N.; Kolmetzky, D.W.; Mangold, A.S.; Roy, R.; Ibetti, J.; Nwokedi, M.; Koch, W.J.; et al. Enhanced NCLX-dependent mitochondrial Ca2+ efflux attenuates pathological remodeling in heart failure. J. Mol. Cell. Cardiol. 2022, 167, 52–66.
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565.
- Martin, J.L.; Costa, A.S.H.; Gruszczyk, A.V.; Beach, T.E.; Allen, F.M.; Prag, H.A.; Hinchy, E.C.; Mahbubani, K.; Hamed, M.; Tronci, L.; et al. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat. Metab. 2019, 1, 966–974.
- Zhang, J.; Wang, Y.T.; Miller, J.H.; Day, M.M.; Munger, J.C.; Brookes, P.S. Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 2018, 23, 2617–2628.
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263.
- Milliken, A.S.; Kulkarni, C.A.; Brookes, P.S. Acid enhancement of ROS generation by complex-I reverse electron transport is balanced by acid inhibition of complex-II: Relevance for tissue reperfusion injury. Redox Biol. 2020, 37, 101733.
- Yin, Z.; Burger, N.; Kula-Alwar, D.; Aksentijevic, D.; Bridges, H.R.; Prag, H.A.; Grba, D.N.; Viscomi, C.; James, A.M.; Mottahedin, A.; et al. Structural basis for a complex I mutation that blocks pathological ROS production. Nat. Commun. 2021, 12, 707.
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435.
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Nikitina, N.N.; Shkurat, T.P.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial mutations associated with cardiac angina. Vessel Plus 2019, 3, 8.
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 8, 166.
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473.
- Hulsmans, M.; Van Dooren, E.; Holvoet, P. Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 264–276.
- Wen, R.; Banik, B.; Pathak, R.K.; Kumar, A.; Kolishetti, N.; Dhar, S. Nanotechnology inspired tools for mitochondrial dysfunction related diseases. Adv. Drug Deliv. Rev. 2016, 99, 52–69.
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420.
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332.
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; Van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res. 2000, 86, 960–966.
- Hu, H.; Lin, Y.; Xu, X.; Lin, S.; Chen, X.; Wang, S. The alterations of mitochondrial DNA in coronary heart disease. Exp. Mol. Pathol. 2020, 114, 104412.
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350.
- Zakirov, F.H.; Zhang, D.; Grechko, A.V.; Wu, W.K.; Poznyak, A.V.; Orekhov, A.N. Lipid-based gene delivery to macrophage mitochondria for atherosclerosis therapy. Pharmacol. Res. Perspect. 2020, 8, e00584.
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541.
- Poredos, P.; Poredos, A.V.; Gregoric, I. Endothelial Dysfunction and Its Clinical Implications. Angiology 2021, 72, 604–615.
- Sima, A.V.; Stancu, C.S.; Simionescu, M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009, 335, 191–203.
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52.
- Moriya, J. Critical roles of inflammation in atherosclerosis. J. Cardiol. 2019, 73, 22–27.
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702.
- Dominic, E.A.; Ramezani, A.; Anker, S.D.; Verma, M.; Mehta, N.; Rao, M. Mitochondrial cytopathies and cardiovascular disease. Heart 2014, 100, 611–618.
- Tretter, L.; Ambrus, A. Measurement of ROS homeostasis in isolated mitochondria. Methods Enzymol. 2014, 547, 199–223.
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G205–G214.
- Frey, R.S.; Gao, X.; Javaid, K.; Siddiqui, S.S.; Rahman, A.; Malik, A.B. Phosphatidylinositol 3-kinase gamma signaling through protein kinase Czeta induces NADPH oxidase-mediated oxidant generation and NF-kappaB activation in endothelial cells. J. Biol. Chem. 2006, 281, 16128–16138.
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88, 179–188.
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal. 2011, 14, 469–487.
- Zhu, Y.; Li, M.; Lu, Y.; Li, J.; Ke, Y.; Yang, J. Ilexgenin A inhibits mitochondrial fission and promote Drp1 degradation by Nrf2-induced PSMB5 in endothelial cells. Drug Dev. Res. 2019, 80, 481–489.
- Ceaser, E.K.; Ramachandran, A.; Levonen, A.L.; Darley-Usmar, V.M. Oxidized low-density lipoprotein and 15-deoxy-delta 12,14-PGJ2 increase mitochondrial complex I activity in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2298–H2308.
- Kinscherf, R.; Deigner, H.P.; Usinger, C.; Pill, J.; Wagner, M.; Kamencic, H.; Hou, D.; Chen, M.; Schmiedt, W.; Schrader, M.; et al. Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: Its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1997, 11, 1317–1328.
- Yang, X.; Pan, W.; Xu, G.; Chen, L. Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 502, 245–254.
- Garza-Lombo, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion 2020, 51, 105–117.
- Dymkowska, D. The involvement of autophagy in the maintenance of endothelial homeostasis: The role of mitochondria. Mitochondrion 2021, 57, 131–147.
- Drabarek, B.; Dymkowska, D.; Szczepanowska, J.; Zablocki, K. TNFalpha affects energy metabolism and stimulates biogenesis of mitochondria in EA.hy926 endothelial cells. Int. J. Biochem. Cell Biol. 2012, 44, 1390–1397.
- Dymkowska, D.; Drabarek, B.; Michalik, A.; Nowak, N.; Zablocki, K. TNFalpha stimulates NO release in EA.hy926 cells by activating the CaMKKbeta-AMPK-eNOS pathway. Int. J. Biochem. Cell Biol. 2019, 106, 57–67.
- Koga, Y.; Akita, Y.; Junko, N.; Yatsuga, S.; Povalko, N.; Fukiyama, R.; Ishii, M.; Matsuishi, T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 2006, 66, 1766–1769.
- Wang, X.L.; Sim, A.S.; Badenhop, R.F.; McCredie, R.M.; Wilcken, D.E. A smoking-dependent risk of coronary artery disease associated with a polymorphism of the endothelial nitric oxide synthase gene. Nat. Med. 1996, 2, 41–45.
- Koga, Y.; Akita, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Tanabe, Y.; Fujimoto, S.; Matsuishi, T. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology 2005, 64, 710–712.