Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mitochondria calcium is a double-edged sword. The low levels of calcium are essential to maintain optimal rates of ATP production, extreme levels of calcium overcoming the mitochondrial calcium retention capacity leads to loss of mitochondrial function. In moderate amounts, ATP synthesis rates are inhibited in a calcium-titratable manner.
- bioenergetics
- calcium overload
- mitochondria
- mitochondrial ultrastructure
- mitochondrial function
1. Introduction: Mitochondrial Calcium—The Good and the Bad
ATP is coupled to nearly every reaction in the body and is necessary for an organism’s survival. This essential energy metabolite is primarily produced by mitochondria in a process known as oxidative phosphorylation. Oxidative phosphorylation is regulated in a manner that ensures the optimal rate of mitochondrial ATP production. ATP breakdown products, ADP, and inorganic phosphate (Pi) are the most potent regulators of oxidative phosphorylation. That said, calcium is also an important regulator but acts as a double–edged sword regarding oxidative phosphorylation [1][2]. Calcium ions enter and leave mitochondria through a variety of specialized channels and transporters in a tissue-specific manner [3]. Moreover, mitochondria possess a unique ability to accumulate massive quantities of calcium in their matrix with devastating consequences [1][2][4][5]. While relatively low amounts of calcium (0 < 40 nmol/mg mitochondria) are essential for energy production, high levels of calcium (>500 nmol/mg) lead to the total collapse of energy homeostasis (Figure 1) [6]. In between, a state known as calcium overload (40–500 nmol/mg), calcium impairs oxidative phosphorylation and presumably contributes to long–term organ dysfunction. These ranges were identified using guinea pig cardiac mitochondria, and external effectors such as cyclosporin A (CsA) can modulate them [2][7]. Thus, the regulation of mitochondrial calcium content is of utmost importance for living tissue.
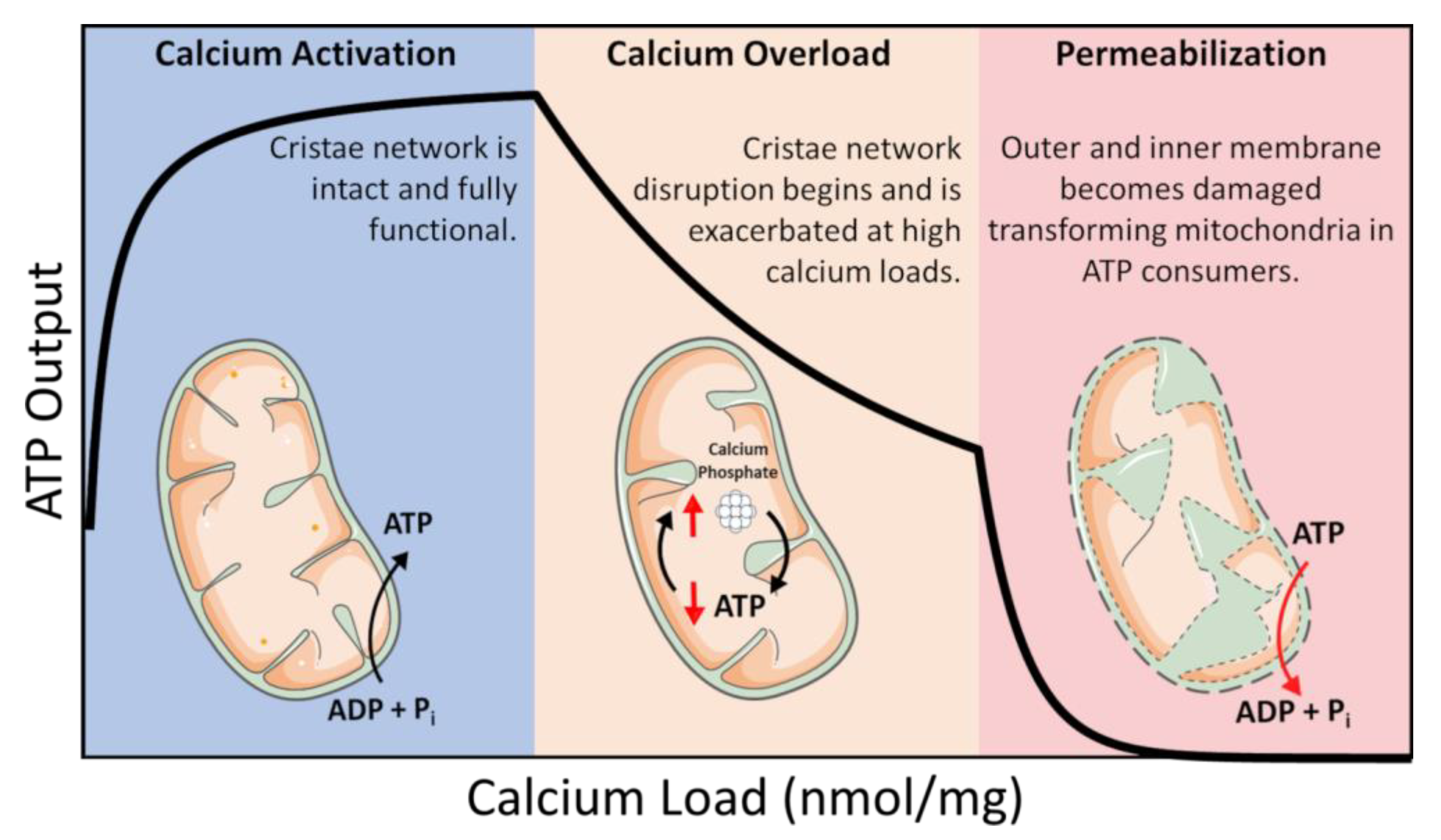
Figure 1. Calcium overload. In low amounts, calcium enhances mitochondrial function by activating several Ca2+–sensitive catabolic enzymes. In moderate amounts, depressed rates of oxidative phosphorylation become observable. In extreme amounts, mitochondria become structurally compromised and consume ATP in a futile attempt to restore homeostasis.
In this intermediate calcium overloaded state, ATP synthesis is inhibited by total mitochondrial calcium in a titratable manner [1][2][8]. This inhibition is relieved after calcium is removed from mitochondria if the total content is below 500 nmol/mg, and calcium removal only partially recovers ATP synthesis capacity at higher loads [5]. Phosphate facilitates mitochondrial calcium uptake but can ultimately lead to cell death via the mitochondrial permeability transition phenomenon [9][10]. While phosphate is thought to act as a permeability transition inducer, it may also serve a dual purpose as a desensitizer under certain conditions [11]. While the molecular details of the mitochondrial permeability transition phenomenon are still debated, the current consensus revolves around the idea of the formation of a proteinaceous pore [12][13]. That said, the conditions required to open this pore in vitro are extreme and would result in irreversible cell death in vivo. Thus, it may not play a significant role in how calcium regulates energy metabolism in living tissue.
2. Calcium Homeostasis, Entry, and Exit Pathways
Mitochondrial calcium homeostasis is primarily regulated by three pathways: the mitochondrial calcium uniporter (MCU), sodium/calcium/lithium exchanger (NCLX), and calcium hydrogen exchanger (CHE). The MCU is the dominant uptake pathway and is comprised of a heteromeric protein complex composed of various subunits including the mitochondrial Ca2+ uptake family (MICU 1, 2, and 3), essential MCU regulator (EMRE), MCU regulator 1 (MCUR1), MCU dominant-negative β–subunit (MCUb), and the solute carrier 25A23 (SLC25A23) [14]. These subunits form a complex on what appears to be an on-demand basis [14][15][16][17]. While an in–depth review on the molecular structure and function of these subunits are beyond the scope of this perspective, researchers would like to refer the readers to references [14][16][17]. Other pathways for Ca2+ uptake including the rapid mode calcium uptake and ryanodine receptors are also speculated to play an important role under certain conditions [18][19][20]. The MCU channel has a high affinity for Ca2+ (Kd ≤ 2 nM); however, it has a low half-activation constant (K0.5~20 mM) [21]. As a result, the MCU is less active at lower concentrations of calcium (0.1–1 µM) and more active at higher extramitochondrial calcium loads [18]. The efflux pathways are primarily controlled by the NCLX and CHE. The NCLX is an electrogenic exchanger that swaps 3 Na+ (or Li+) for 1 Ca2+ [22][23]. Consequently, this reaction is electrogenic and sensitive to the mitochondrial membrane potential. Under physiological conditions, the CHE swaps calcium out for protons in the matrix in a manner that is presumed to be an electroneutral ratio of 1 Ca2+ per 2 H+ [24]. The calcium hydrogen exchanger functions independently of sodium and is present at much lower activities in tissues with high energy demand [25]. For instance, the CHE is dominant in the liver and other relatively quiescent tissues, while NCLX is predominant in the heart, brain, and other high activity tissues [26][27][28][29][30][31].
Under basal conditions, cytosolic Ca2+ concentrations are maintained in the 100 nM range [32]. For cardiomyocytes and skeletal myocytes, the range of global or average cytosolic Ca2+ concentrations during peak contraction that mitochondria are exposed to fall within 1 µM but can peak two to three times higher under stimulatory conditions [33]. That said, some mitochondria are exposed to higher concentrations (~10–100 µM) in microdomains associated with mitochondrial–SR contact sites [34][35][36]. In either the basal or stimulatory condition, intramitochondrial Ca2+ levels remain low as long as mitochondria remain coupled [34]. This form of calcium regulation is attributed to the mitochondrial calcium buffering system. What makes mitochondria particularly relevant in this scenario is their ability to store large amounts of calcium in their matrix [37].
This is of relevance in high–energy demand tissue as the mitochondrial membrane potential, Ca2+, and Na+ are the main regulators of mitochondrial calcium homeostasis. Additionally, while Ca2+ uptake is very sensitive to changes in membrane potential, Ca2+ efflux is less sensitive [38]. As a result, the MCU channel can load Ca2+ into the matrix at a rate far exceeding the NCLX matrix calcium clearance (1400 and 20 nmol Ca2+ min−1 mg mitochondrial protein−1, respectively) [39]. Hence, under conditions that disrupt cytosolic calcium homeostasis, Ca2+ uptake through MCU floods the matrix of energized mitochondria with massive amounts of Ca2+ [8]. Left unchecked, membrane potential loss precipitates a catastrophic collapse in energy homeostasis [2][40]. This phenomenon is often ascribed to the permeability transition phenomenon and has detrimental consequences for cell health and longevity [4][9][39][41][42]. This scenario places mitochondria in a vulnerable position, leading some to view the permeability transition phenomenon as a calcium overload release valve [43][44]. Regardless, the mitochondria Ca2+ uptake and removal processes are highly regulated with compensatory mechanisms in place to ensure cellular homeostasis and survivability. When such regulatory mechanisms fail, mitochondria become overloaded with calcium, and energy homeostasis collapses.
3. Calcium TCA and ETC
Calcium also influences mechanisms driving mitochondrial energy production and metabolic activity. For example, calcium regulates the activity of metabolic enzymes in the TCA cycle including pyruvate dehydrogenase, isocitrate dehydrogenase, and α–ketoglutarate dehydrogenase [2]. The TCA cycle generates reducing equivalents (such as NADH and UQH2) used by the proton pumps that establish the membrane potential and alkalize the mitochondrial matrix relative to the cytoplasm [23]. This, in turn, biases ATP synthase away from its more favorable ATP hydrolysis set point to an operating regime conducive for ATP production via oxidative phosphorylation. Therefore, in high–energy demanding tissue where cells are constantly exposed to transient Ca2+ signals, calcium homeostasis is intrinsically linked to ATP production both through the TCA cycle and oxidative phosphorylation (oxphos) to regulate cellular bioenergetics.
Under most conditions, the membrane potential is the primary determinant of the ratio of matrix ATP to ADP and inorganic phosphate (i.e., the matrix ATP/ADP/Pi ratio). As Ca2+ is injected into mitochondria, matrix ATP/ADP ratios decline, the membrane potential depolarizes to a degree, and matrix pH increases [42][45]. As the matrix pH becomes more basic, dihydrogen phosphate (H2PO4−) is effectively driven into the mitochondria in symport with H+ in an electroneutral fashion. The phosphate carrier facilitates his symport, and when H2PO4− enters the matrix, it undergoes a deprotonation event and forms HPO42− [46]. Under these conditions, elevated Pi levels in the matrix facilitate the formation of calcium phosphate complexes. The formation of these complexes involves the deprotonation of HPO42− into the phosphate trianion (PO43−) further releasing a H+. This helps counteract the significant alkalizing effect of accelerated proton pumping and charge replacement caused by Ca2+ uptake. Overall, a slight depolarization will alkalize the matrix pH which has the net effect of enhancing mitochondrial Ca2+ sequestration [40]. However, when the current generated by Ca2+ uptake exceeds the proton pumping current, thermodynamic driving forces reverse the F1FO ATP synthase activity and pumps protons out of the matrix via ATP hydrolysis [6]. When ATP is hydrolyzed from this reversal, the phosphate released can participate in phosphate precipitate formation until ATP is exhausted and the metabolic system collapses [37][47]. This is just one possible scenario as ATP hydrolysis is not a required source of inorganic phosphate during precipitate formation. In the presence of oligomycin, ATP synthase is inhibited, and mitochondria still possess the ability to take up massive amounts of Ca2+ when sufficient Pi is present [48]. Ultimately, when matrix Ca2+ concentrations exceed a threshold level, the formation of calcium phosphate precipitates in the matrix has the effect of reducing the mitochondrial free Ca2+ levels to manageable amounts via a type of buffering mechanism but at the expense of oxidative phosphorylation capacity [1][18].
4. Mitochondrial Calcium Buffering
Researchers know that the consequences of precipitate formation operate on a spectrum (Figure 1), but they do not fully comprehend the mechanism. At low concentrations, calcium phosphate precipitates can have a protective effect. Whereas at high concentrations, precipitates can destabilize the mitochondrial cristae network [2][5]. This has been confirmed by others in which mitochondria loaded with Ca2+ resulted in calcium phosphate precipitates occupying more than 20% of the matrix volume [2]. One potential mechanism that leads to metabolic dysfunction is that precipitates may mechanically destabilize membrane structures by disrupting proteins involved in maintaining the cristae structure [5]. Another idea is that precipitates may serve as physical barriers limiting metabolite and substrate diffusion across the matrix [1]. The regulatory characteristics of the phosphate precipitation buffering mechanism remain enigmatic, but one concept boils down precipitate formation to a simple thermodynamic argument [37][47]. A second concept includes the idea that precipitate formation requires nucleation sites [49]. Full occupancy of these sites might dampen the extent of phosphate buffering within the mitochondrial matrix and send free Ca2+ high into pathological concentrations. The unknown nature of these potential nucleation sites makes it challenging to devise effective genetic and pharmacological approaches to manipulate mitochondrial calcium buffering. Thus, further study is required to resolve some of these unknowns.
This entry is adapted from the peer-reviewed paper 10.3390/biom12121891
References
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719.
- Strubbe-Rivera, J.O.; Chen, J.; West, B.A.; Parent, K.N.; Wei, G.W.; Bazil, J.N. Modeling the Effects of Calcium Overload on Mitochondrial Ultrastructural Remodeling. Appl. Sci. 2021, 11, 2071.
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68.
- Petronilli, V.; Nicolli, A.; Costantini, P.; Colonna, R.; Bernardi, P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim. et Biophys. Acta 1994, 1187, 255–259.
- Strubbe-Rivera, J.O.; Schrad, J.R.; Pavlov, E.V.; Conway, J.F.; Parent, K.N.; Bazil, J.N. The mitochondrial permeability transition phenomenon elucidated by cryo-EM reveals the genuine impact of calcium overload on mitochondrial structure and function. Sci. Rep. 2021, 11, 1–15.
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973.
- Duong, Q.V.; Hoffman, A.; Zhong, K.; Dessinger, M.J.; Zhang, Y.; Bazil, J.N. Calcium overload decreases net free radical emission in cardiac mitochondria. Mitochondrion 2020, 51, 126–139.
- Carafoli, E. The fateful encounter of mitochondria with calcium: How did it happen? Biochim. Biophys. Acta 2010, 1797, 595–606.
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125.
- Petronilli, V.; Cola, C.; Massari, S.; Colonna, R.; Bernardi, P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J. Biol. Chem. 1993, 268, 21939–21945.
- Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P. Phosphate Is Essential for Inhibition of the Mitochondrial Permeability Transition Pore by Cyclosporin A and by Cyclophilin D Ablation. J. Biol. Chem. 2008, 283, 26307–26311.
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J Mol Cell Cardiol. 2020, 144, A3–A13.
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 1–12.
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. Physiol. 2021, 320, C465–C482.
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.d.l.F.; Bogorad, R.; et al. MICU1 Controls Both the Threshold and Cooperative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013, 17, 976–987.
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607.
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. et Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2006–2011.
- Wei, A.-C.; Liu, T.; Winslow, R.L.; O’Rourke, B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: Two components of Ca2+ uptake and role of phosphate buffering. J. Gen. Physiol. 2012, 139, 465–478.
- Mishra, J.; Jhun, B.S.; Hurst, S.; Csordás, G.; Sheu, S.S. The Mitochondrial Ca2+ Uniporter: Structure, Function, and Pharmacology. Handb Exp Pharm. 2017, 240, 129–156.
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102.
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364.
- Garbincius, J.F.; Elrod, J.W. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992.
- Dash, R.K.; Beard, D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008, 586, 3267–3285.
- Austin, S.; Mekis, R.; Mohammed, S.E.M.; Scalise, M.; Pfeiffer, C.; Galluccio, M.; Borovec, T.; Parapatics, K.; Vitko, D.; Dinhopl, N.; et al. MICSI is the Ca2+/H+ antiporter of mammalian mitochondria. BioRxiv 2021, 1–53.
- Gunter, K.K.; Zuscik, M.J.; Gunter, T.E. The Na(+)-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J. Biol. Chem. 1991, 266, 21640–21648.
- Wingrove, E.D.; Gunter, E.T. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 1986, 261, 15166–15171.
- Wingrove, E.D.; Gunter, E.T. Kinetics of mitochondrial calcium transport. I. Characteristics of the sodium-independent calcium efflux mechanism of liver mitochondria. J. Biol. Chem. 1986, 261, 15159–15165.
- Garbincius, J.F.; Luongo, T.S.; Jadiya, P.; Hildebrand, A.N.; Kolmetzky, D.W.; Mangold, A.S.; Roy, R.; Ibetti, J.; Nwokedi, M.; Koch, W.J.; et al. Enhanced NCLX-dependent mitochondrial Ca2+ efflux attenuates pathological remodeling in heart failure. J. Mol. Cell. Cardiol. 2022, 167, 52–66.
- Drago, I.; Pizzo, P.; Pozzan, T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011, 30, 4119–4125.
- Pizzo, P.; Drago, I.; Filadi, R.; Pozzan, T. Mitochondrial Ca²⁺ homeostasis: Mechanism, role, and tissue specificities. Pflug. Arch 2012, 464, 3–17.
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408.
- Bers, D.M. Altered Cardiac Myocyte Ca Regulation In Heart Failure. Physiology 2006, 21, 380–387.
- Hale, C.C.; Bossuyt, J.; Hill, C.K.; Price, E.M.; Schulze, D.H.; Lederer, J.W.; Poljak, R.; Braden, B.C. Sodium-Calcium Exchange Crystallization. Ann. N. Y. Acad. Sci. 2006, 976, 100–102.
- Boyman, L.; Lederer, W.J. How the mitochondrial calcium uniporter complex (MCUcx) works. Proc. Natl. Acad. Sci. USA 2020, 117, 22634–22636.
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486.
- Szanda, G.; Koncz, P.; Várnai, P.; Spät, A. Mitochondrial Ca2+ uptake with and without the formation of high-Ca2+ microdomains. Cell Calcium 2006, 40, 527–537.
- Chalmers, S.; Nicholls, D.G. The Relationship between Free and Total Calcium Concentrations in the Matrix of Liver and Brain Mitochondria. J. Biol. Chem. 2003, 278, 19062–19070.
- Zhang, X.; Tomar, N.; Kandel, S.M.; Audi, S.H.; Cowley, A.W.; Dash, R.K. Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney. Cells 2021, 11, 131.
- Bernardi, P.; Petronilli, V. The permeability transition pore as a mitochondrial calcium release channel: A critical appraisal. J. Bioenerg. Biomembr. 1996, 28, 131–138.
- Chinopoulos, C.; Adam-Vizi, V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010, 277, 3637–3651.
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293.
- Bernardi, P. Mitochondrial Transport of Cations: Channels, Exchangers, and Permeability Transition. Physiol. Rev. 1999, 79, 1127–1155.
- Wu, N.; Li, W.-N.; Shu, W.-Q.; Lv, Y.; Jia, D.-L. Blocking the mitochondrial permeability transition pore with cyclosporine-A can restore cardioprotection of ischemic postconditioning in hypercholesterolemic rat heart. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 446–454.
- Bernardi, P. Mechanisms for Ca2+-dependent permeability transition in mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 2743–2744.
- Poburko, D.; Domingo, J.S.; Demaurex, N. Dynamic Regulation of the Mitochondrial Proton Gradient during Cytosolic Calcium Elevations. J. Biol. Chem. 2011, 286, 11672–11684.
- Seifert, E.L.; Ligeti, E.; Mayr, J.A.; Sondheimer, N.; Hajnóczky, G. The mitochondrial phosphate carrier: Role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 2015, 464, 369–375.
- Nicholls, D.G.; Chalmers, S. The Integration of Mitochondrial Calcium Transport and Storage. J. Bioenerg. Biomembr. 2004, 36, 277–281.
- Carafoli, E.; Patriarca, P.; Rossi, C.S. A comparative study of the role of mitochondria and the sarcoplasmic reticulum in the uptake and release of Ca++ by the rat diaphragm. J. Cell. Physiol. 1969, 74, 17–29.
- Duvvuri, B.; Lood, C. Mitochondrial Calcification. Immunometabolism 2021, 3, 8.
This entry is offline, you can click here to edit this entry!