Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The ubiquitin-26S proteasome system and autophagy are two major protein degradation machineries encoded in all eukaryotic organisms. While the ubiquitin-26S proteasome system (UPS) is responsible for the turnover of short-lived and/or soluble misfolded proteins under normal growth conditions, the autophagy-lysosomal/vacuolar protein degradation machinery is activated under stress conditions to remove long-lived proteins in the forms of aggregates, either soluble or insoluble, in the cytoplasm and damaged organelles.
- ubiquitin
- proteasome
- autophagy
- ubiquitylation
- protein degradation
- liquid–liquid phase separation
1. Structure and Activity Control of the 26S Proteasome
The 26S proteasome is one of the largest multi-subunit protein complexes that is composed of two functionally distinguished subcomplexes (Figure 1). The proteolytic part is attributed to a barrel-shaped 20S core particle (CP) that comprises four axially stacked heteroheptameric rings (two outer α- and two inner β-rings) [1]. Among the four rings, three of the seven pairs of the β subunits (β1, β2, and β5) possess six catalytic sites that have caspase-like (cleave after acidic amino acid), trypsin-like (post-basic), and chymotrypsin-like (post-hydrophobic) specificities, giving the proteolytic function of the 26S proteasome [2]. These subunits utilize the hydroxyl group of the terminal threonine residue as the catalytic nucleophile for attacking peptide bonds of a substrate [3]. However, the 20S CP alone does not have the proteolytic function, because the N-termini of the seven pairs of the α-ring subunits completely blocks the accessibility of unregulated substrates to the proteolytic chamber by sealing the entrance pore (“gate”) on both sides of the internal β-rings [4][5][6][7].
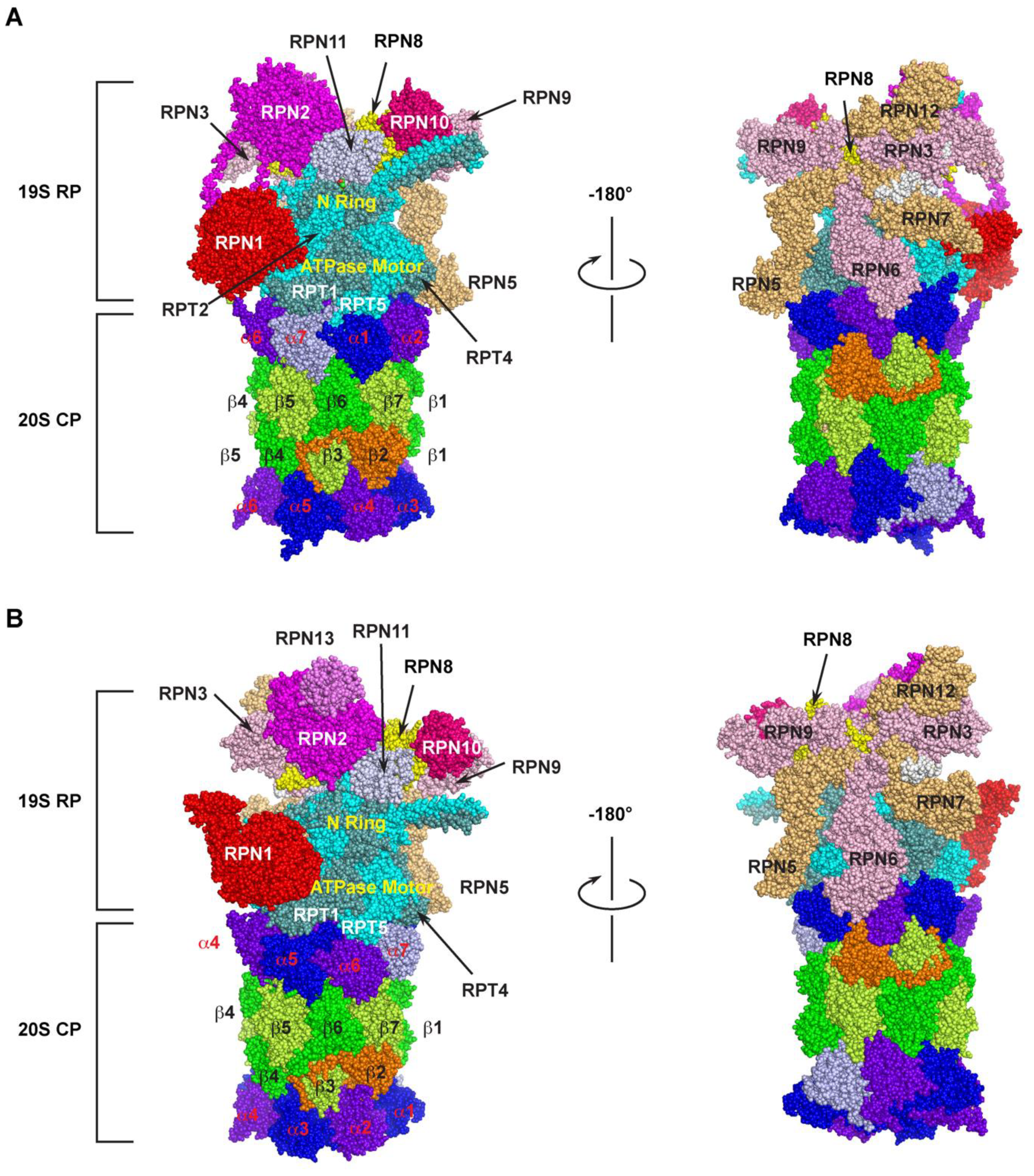
Figure 1. Cryo-electron microscopy (Cryo-EM) structure of the 26S proteasome. The spherical structure is generated based on PDB 6MSK [8] and 3JCP [9] for humans (A) and yeast (B) 26S proteasome, respectively. Two mirror images were shown for the detailed organization of base and lid subunits of the 19S RP subcomplex in each proteasome. The parts of N-ring and ATPase motor ring of RPT hexameric complex are indicated. The subunits of α-ring and β-ring of the 20S CP are highlighted with different colors. The human 26S proteasome structure does not include RPN13.
The 19S regulatory particle (RP) is designed to recognize the substrate, remove ubiquitin modifications, control the opening of the substrate entrance pore, unfold, and translocate the substrate into the 20S core. It binds to either or both ends of the 20S CP to assemble a 26S or 30S proteasome holo-complex, respectively, based on their sedimentation coefficients [10][11]. According to organization and function, a typical RP can be further divided into a base and a lid subcomplex [12][13][14].
The base is made up of 10 subunits, including four Regulatory Particle Non-ATPase (RPN)1 [15], RPN2 [16][17], RPN10 [18], and RPN13 [19], and a hexameric ATPase motor ring composed of six Regulatory Particle Triple-A ATPases (RPT) 1–6 [20][21] (Figure 1). In addition to the ATP hydrolysis function of the Triple-A ATPases active domain, the RPT subunits carry an N-terminal oligonucleotide/oligosaccharide-binding (OB)-fold domain [22][23][24] and a C-terminal HbYX motif (where Hb stands for a hydrophobic residue; Y for tyrosine; and X for any amino acid) [25]. The OB domains form a rigid N-terminal ring that is stacked on top of the AAA-domain ring [15][21][23]. Upon recognition, a ubiquitylation substrate needs to go through the N-ring before engaging with the AAA-ATPases that convert ATP hydrolysis energy into mechanical pulling for its unfolding and translocation into the 20S core [24][26][27]. It was shown that an unstructured initiation region with at least 20–25 amino acids is required for a substrate to be engaged with the ATPase motor [24][28][29][30][31][32][33][34]. This necessity may provide the second degradation code for a ubiquitylation substrate in addition to the topology of polyubiquitin chains. The C-terminal HbYX motif of an RPT subunit binds to a pocket between the 20S CPs α subunits functioning as a ‘‘key in a lock’’ to induce gate opening and allow substrate entry [25][35][36]. Two of the four RPN subunits, RPN10 and RPN13, function as the receptors of ubiquitylation substrates [19][20]. RPN1 and RPN2 are the two paralogues with the largest size among the entire group of the 26S subunits [37][38]. Their central α-turn-α proteasome/cyclosome (PC) motifs form a toroid structure that is further extended by their divergent flexible N- and C-terminal regions. The large size and the toroid structure give them a role in functioning as flexible scaffolds between the base and the lid [17][39][40][41] (Figure 1A). The different positions of RPN1 and RPN2 within the 19S RP also fulfil their specific functions. For example, RPN1 also functions as the third receptor for the poly-ubiquitin chain to facilitate deubiquitylation [15][20][37], while RPN2 tightly interacts with RPN13 and a proteasome-associated DUB, UCH37/UCH-L5 [42][43][44] (Figure 1B). RPN2 was also shown to bind with importin αβ for mediating nuclear imports of proteasomal components [45].
The RP lid contains nine subunits that include RPN3, RPN5-9, RPN11-12, and RPN15 [46] (Figure 1). Among them, RPN11 is a Zn2+-dependent deubiquitylase (DUB) [21][47][48], forming a hetero-dimeric complex with RPN8 [49][50]. The six Proteasome–CSN–eIF3 (PCI) domains containing subunits, RPN3, 5, 6, 7, 9, and 12, assemble a horseshoe structure playing a scaffolding function to facilitate inter-subunit binding [13][14][38] (Figure 1A,B, right panel). RPN8 sits in the horseshoe through interacting with RPN3 and RPN9. It also forms a heterodimer with RPN11, projecting the active site of RPN11 near the N-ring of RPTs (Figure 1A-B, left panel). The C-terminus of RPN3 was shown to contact the N-ring of the RPT base proposed to form a composite active site for substrate deubiquitylation and unfolding [38][50]. Thus, the C-terminus of RPN3 may function as a sensor of substrates engaged in the N-ring for initiating conformational changes of the lid to activate RPN11 and the composite active site [38][51].
2. Composition of the Plant 26S Proteasome
The past decade of structural studies have shed rich insights into the dynamic function of substrate recognition, engagement, and degradation in a functional proteasome of yeast [13][24][51][52][53][54][55][56], humans [8], and mammals [57]. Unfortunately, no structural information has been available for a plant proteasome, although it has been clearly demonstrated that the UPS plays a tremendous importance in plant evolution, growth, and development [58][59][60][61][62][63]. The lack of such a critical study in understanding the kinetics and structure of the major degradation machinery in plants is reminiscent of a continuing issue of the weak attention on recognizing plant science as an important discipline for discoveries in basic biology [64]. Nevertheless, the pioneering work conducted in the Vierstra lab has uncovered multiple conserved and specific mechanisms of plant proteasomes, including the first discovery of proteaphagy (see below).
Through early genomic studies, 23 genes encoding a complete list of 14 20S CP proteasome subunits [65], 11 genes for RPT1-6 [66], and 6 genes for non-ATPase subunits (RPN1, 2, 6, 8, 10, and 11) [67], were identified in Arabidopsis, primarily in the representative Columbia-0 accession. In addition to gene identification, genomic DNA analysis by Southern blotting confirmed gene duplications for multiple CP and RP genes [65][66]. The biochemical function of some gene products, including three CP genes, PAC1 (α3), PAE1(α5), and PBC2 (β3) [65], and five RPT genes, RPT1 and 3–6, were confirmed by complementation assay in yeast that lacked the expression of a corresponding orthologous gene. However, the big picture about the Arabidopsis holo-proteasome complex was lacking by this assay, due to an incomplete genome sequence database and the unavailability of comprehensive evolutionary studies.
Using an improved bioinformatic analysis pipeline, the researchers were able to find genes de novo for 30 out of all 34 proteasome subunits in seven flowering plants that include two evolutionary distant genera, Oryza and Arabidopsis, each with three species, and the basal flowering plant, Amborella trichopoda [68]. RPN11, 12, 13, and 15 were not studied because the simplicity of their domain structures was not helpful for distinguishing them from members of the eIF3 and CSN complexes. Since the Oryza sativa Nipponbare reference genome represents the second best-annotated plant genome [69], the failure in finding an RPN8 orthologue in the rice genome is not likely due to genome sequence errors. The researchers also discovered that most proteasome members from the two genera form an independent subclade with strong statistical significance support, further suggesting the functional diversification of plant proteasomes [68] (Figure 2). How this evolutionary divergence contributes to the biochemical and functional specialization of proteasomes requires further investigation.
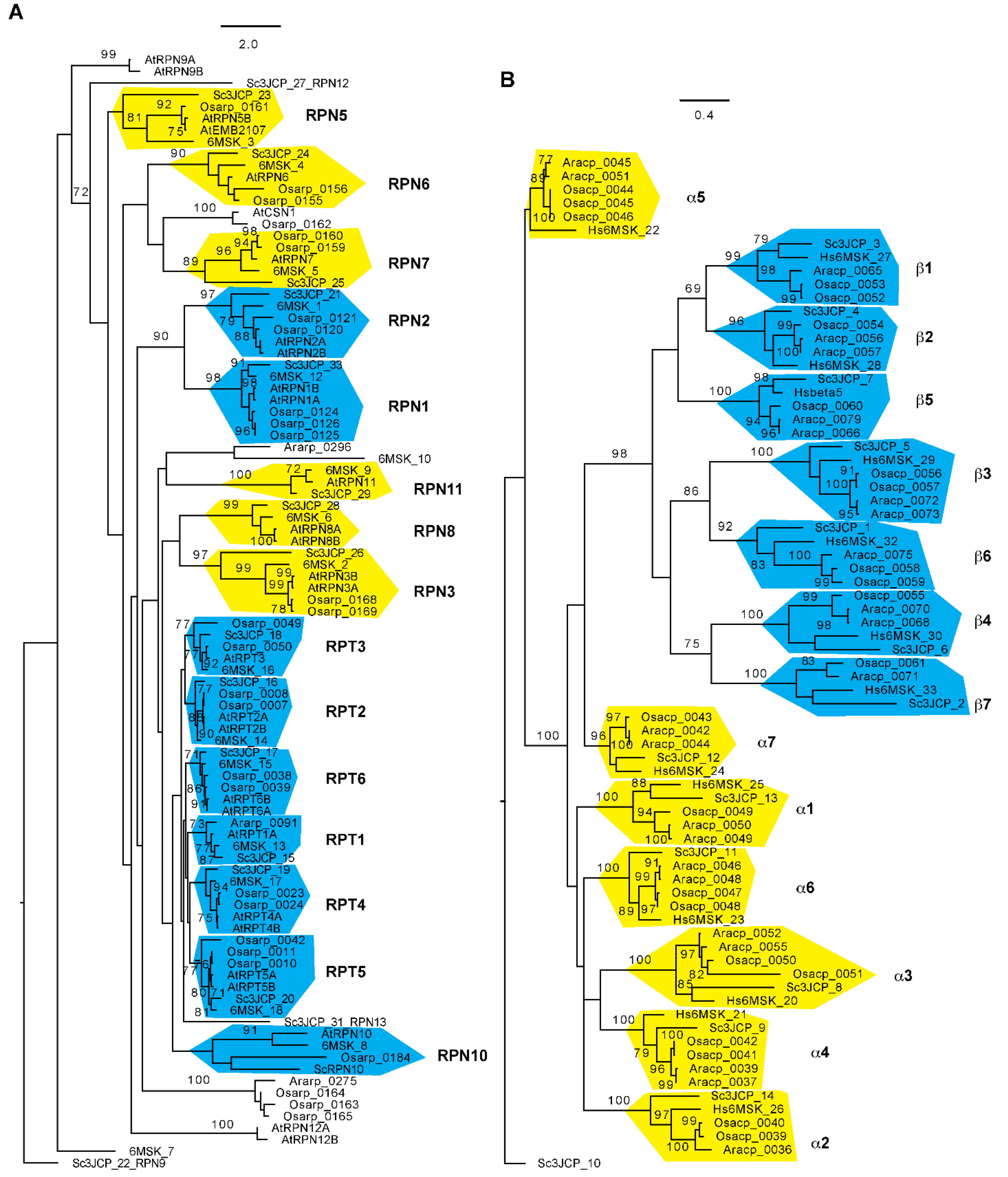
Figure 2. Phylogenetic relationship of 26S proteasome subunits among Arabidopsis, rice, yeast, and humans. The sequences and sequence identifications of Arabidopsis and rice 26S proteasome subunits were retrieved from Hua and Yu, 2019 [68], and those for yeast and human 26S proteasomes were obtained from Luan et al. (2016) [9] and Dong et al. (2019) [8], respectively. The RP (A) and CP (B) phylogenetic trees were separately generated using RAxML (Version 8.1) with the PROTGAMMAJTT substitution model [70]. A significant clade is indicated with a bootstrap value greater than 70% that was calculated based on 1000 replications. Independent clades are labeled and shaded with yellow ((A): lid subunits; (B): α subunits) or cyan ((A): base subunits; (B): β subunits) color. Scale bar: average substitutions per site. Ara: Arabidopsis; Osa: Oryza sativa; Hs: Homo sapiens; Sc: Saccharomyces cerevisiae.
It was a breakthrough discovery in proteasome biochemistry studies to functionally replace a proteasome subunit with a tagged recombinant version [71]. This replacement not only allows for the affinity purification of proteome complexes in vivo for proteomics studies, but also develops proteasomal reporters for monitoring intracellular dynamics of proteasomes through live cell imaging. For example, the Vierstra lab has developed three different Arabidopsis transgenic lines, in which the endogenous CP subunit PAG1 (α7) and two isoforms of the RP subunit RPT4 were replaced with FLAG-tagged fusions. Using a C-terminal tagged PAG1-FLAG in conjugation with in-depth mass spectrometry proteomic sequencing, the two duplicated isoforms of most 26S subunits were identified, suggesting the presence of a heterogeneous population of 26S proteasome particles in Arabidopsis [72]. This raised an intriguing question as to whether the plant proteasomes are assembled in an isoform-specific manner. To address this question, the group cleverly designed two N-terminal FLAG-tagged RPT4 isoforms to develop FALG-RPT4a rpt4a-1 and FALG-RPT4b rpt4b-2 transgenic Arabidopsis plants. Utilizing these two transgenic plants and a similar affinity purification-based mass spectrometry proteomics, the authors failed to identify 26S proteasome isotypes that are specific to the RPT4 paralogues. This seems to be contradictory to the case of mammalian proteasomes, in which different paralogues of β1, β2, and β5 subunits were found in specialized proteasome isoforms with varied proteolytic activities, such as immunoproteasomes and thymoproteasome [73][74]. Since these specialized mammalian proteasomes are present in a tissue, organ, and physiology-specific manner [73], the failure of discovering paralogue-specific 26S proteasome isotypes in Arabidopsis seedlings does not mean that there is an absence of such types of proteasomes in plants. Indeed, the researchers recently discovered that the proteasomes in seedlings and developing siliques possess different catalytic activities and sensitivities to the inhibition of MG132, strongly indicating their presence in plants [75]. Nevertheless, such an affinity for purification-based proteomic studies have discovered the complete set of Arabidopsis proteasome subunits that are orthologous to the yeast and mammalian counter partners as well as a rich group of proteasome-associated proteins, including proteasome assembly chaperons, a PA200/Blm10 regulator that caps the CP, and a ubiquitin-binding shuttle factor DSK2 [72][76].
3. Post-Assembly Regulation of Proteasomes
Due to the space limit, this entry does not focus on the dynamic synthesis of a 26S proteasome complex. For this topic, the readers are referred to recent papers written by Marshall and Vierstra [77], Yedidi et al. [78], Enenkel [10], Mao [79], Budenholzer et al. [80], and Kato and Satoh [81]. Given the large composition, it is energetically costive to assemble and maintain the equal stoichiometry of a minimum of 34 proteasome subunits in a holo-complex (Figure 1 and Figure 2). The requirements of a high-energy investment can inevitably result in errors in both assembling the complete complex and maintaining the functionality of the holo-complex when unfavorable conditions occur. Such stress conditions include, but are not limited to, nutrient starvations, oxidative damages, inhibitory effects, defective subunits resulting either from genetic mutations or translational errors, and subunit misassembly [82]. In this section, the researchers discuss how post-assembled proteasomes with different biophysical states are regulated to meet the requirements of cellular growth and development.
3.1. Proteasome Storage Granules (PSGs)
Through labelling a proteasome subunit with a fluorescent marker, such as a green fluorescent protein (GFP), it is possible to monitor the dynamic localization of a proteasome. For example, most yeast proteasomal subunits can be chromosomally replaced with a GFP-tagged variant. Using this strategy, it was shown that ~80% of the 26S proteasomes were nucleus-localized in actively dividing yeast and mammalian cells [10][83][84]. The nuclear presence of the GFP-labeled proteasome maturation factor Ump1 suggests that some particles represent 20S CP precursor complexes that are in the process of maturation in the nucleus. Thus, the major proportion of both mature and immature proteasomes are present in the nucleus.
Cells are growing under a dynamic shifting environment, which are in many cases suboptimal or even unfavorable. These stress environmental signals can trigger different responsive programs to reprogram intracellular metabolism. The proteasome activities are unequivocally regulated by these signals. Upon carbon starvation, yeast cells enter quiescence [85]. Along with a large fraction of metabolic proteins forming reversible macroscopic foci in quiescent cells [86], nucleus-localized proteasomes were also discovered to dissociate into CP and RP particles to be exported into the cytoplasm to form so-called proteasome storage granules (PSGs) [87]. It was shown that both CP and RP are required to form PSGs [88][89]. However, no holo-complexes are assembled due to declined ATP levels [90]. Thus, PSGs are inactive. In yeast, CP is found to be associated with Blm10, which may also prevent its reassembly with RP [91].
Through high-throughput microscopy on a collection of yeast null-mutants in combination with proteomic studies, the yeast PSGs were discovered to contain not only disassembled CP and RP particles, but also a significant proportion of free ubiquitin molecules that are essential for the nuclear export and sequestration of proteasomes into PSGs [92]. Because all components in the PSGs can revert their biological functions upon the onset of cell proliferation or growth, the PSGs are considered as reservoirs for both proteasome and ubiquitin, for cells to survive under an austerity budget of energy [92]. PSGs represent one type of protein droplets formed through a process now widely recognized as liquid–liquid phase separation (LLPS) [93]. However, because LLPS often requires the presence of intrinsically disordered protein–protein interaction domains that are rare in proteasome subunits [94], the detailed mechanism in the formation of PSGs remain an enigma [95]. It is also unclear whether plants utilize PSGs as a defensive mechanism to overcome carbon starvation. Plant proteasomes seem to be equally localized in both the nucleus and cytoplasm for actively growing seedlings [96][97]. Whether and how the CP and RP subcomplexes in the nucleus and cytoplasm are dissembled simultaneously and intermingled into the cytoplasmic PSGs remains largely unknown.
3.2. Proteaphagy
Unlike carbon starvation, nitrogen starvation triggers the autophagy-mediated vacuolar and lysosomal degradation of proteasomes in plants/yeast and mammalian cells, respectively [97][98][99]. It was also found in plants that the treatment of a tri-peptide aldehyde proteasome inhibitor, carbobenzoxy-Leu-Leu-leucinal (MG132), induced the degradation of the holo-proteasome complex that requires the intact autophagic activity [97]. The discovery of nutrient starvation, genetic aberration, and chemical inhibition-induced autophagic degradation of whole proteasomes in plants led to the term “proteaphagy”, first coined by Marshall et al. [97]. Further studies from the group found that nitrogen depletion-induced proteaphagy differed from that induced by MG132 inhibition. While the authors speculated that nitrogen depletion-induced proteaphagy is regulated through a general bulk autophagy pathway, they uncovered RPN10 as a new selective receptor that specifically bridges the MG132-inhibited proteasomes into autophagosome through interaction with both heavily ubiquitylated proteasomes and AuTophaGy (ATG)8 [97]. The discovery of proteaphagy provides new evidence showing the integration of the two major protein degradation pathways in PQC [100].
However, many unaddressed questions remain. For example, the nitrogen depletion-induced plant proteaphagy is independent on known autophagy cargo receptors, including next to BRCA1 (NBR1) and RPN10, which is suggestive of a general autophagy process [97]. However, P62, a receptor binding both polyubiquitin chains and microtubule-associated protein 1 light chain 3 (LC3; the mammalian ATG8 orthologue) proteins, is required for amino acid starvation-induced proteaphagy in mammalian cells [98]. Through both pharmacological treatment and nitrogen starvation assays, proteaphagy in yeast was also discovered to be selective and it required factors not involved in general autophagy [99]. Plant proteaphagy can be hijacked by bacteria for enhancing virulence through degrading proteasomes. This type of proteaphagy was shown to be induced by a type III effector, HopM1, from bacterial cells, and was activated in a manner comparable to MG132 inhibition. However, whether RPN10 functions as a receptor is yet unclear. Interestingly, NBR1-dependent selective autophagy, albeit activated by bacterial infection, counteracts disease progression. The authors suggested that NBR1 contributes to the turnover of ubiquitylated substrates that were hyperaccumulated during bacterial infection [101]. Plant proteaphagy can also be activated developmentally. Through monitoring the daily changes of proteasome subunits, ubiquitylated proteins and autophagy activities in developing siliques upon pollination for an 8-day developmental period, the researchers recently discovered a relay model of the two degradation pathways in regulating silique/seed development in Arabidopsis. The researchers uncovered that proteaphagy is activated when late heart embryos are developed. Given the same developmental trend of silique proteaphagy in wild types and the rpn10-1 mutant (expressing a truncated RPN10 unable to bind ubiquitylated substrates), the silique proteaphagy is not likely to be selected by RPN10 either. Whether this is due to a general bulk autophagy degradation or NBR1-dependent selective autophagy requires further investigation [75].
The detailed upstream signaling of proteaphagy is not clear either. For example, why do nitrogen depletion and carbon starvation result in different outcomes of proteasome regulation, although both activate autophagy pathways through the master nutrient sensor TORC1? The former induces proteasome degradation through proteaphagy [97][98][102], while the latter prevents the elimination of proteasomes through PSGs [87]. Interestingly, proteaphagy upon nitrogen starvation in yeast involves CP and RP dissociation, nuclear export, and the independent vacuolar targeting of CP and RP subcomplexes. The inhibition of proteaphagy leaves most RPs in the nucleus but discharges CPs into cytosol either as free forms or to be localized into the granular structures within the cytosol [102]. Since the formation of PSGs requires both CP and RP complexes [88][89], can these granular structures be PSGs formed upon carbon starvation? It is yet unknown whether nitrogen starvation, like amino acid starvation in mammalian cells, enhances the ubiquitylation of proteasomes in yeast and plants. Affinity-purified proteasomes in seedlings treated with or without nitrogen starvation did not result in any significant changes of proteasome ubiquitylation [97]. Does this difference result from sequence divergence of proteasome subunits across different species and kingdoms?
3.3. Aggrephagy
Misfolded or malfunctioning proteins may coalesce into perinuclear and MTOC-localized aggresomes through dynein-dependent retrograde transport on microtubes in mammalian cells [103]. In addition, they have also been found in both yeast and mammalian cells to form two other MTOC-independent aggresome-like structures, a soluble juxtanuclear quality control (JUNQ) compartment and an insoluble protein deposit (IPOD) compartment [104]. Since misfolded proteins are cytotoxic, sequestering them into proteinaceous compartments not only provides cytoprotective functions, but also accelerates their clearance by activating UPS and/or autophagy-lysosome/vacuole degradation machineries [103][104][105]. Clearance of cytosolic aggregated by autophagy-lysosomal/vacuolar degradation is generally termed as aggrephagy [105].
Impaired proteasomes by either MG132 reverse inhibition or partially genetic mutations have been found in mammalian aggresomes and yeast IPOD droplets, respectively. Both inclusions were targeted by autophagy-mediated lysosomal or vacuolar degradations [106][107]. The formation of proteasome-containing aggresomes and IPOD droplets was verified by the requirements of HDAC6- and dynein-mediated transport and the colocalization with IPOD markers (Hsp104 and Rnq1), respectively [106][107]. Although JUNQ and IPOD were initially defined by the presence and absence of ubiquitylated proteins, respectively [104], ubiquitylated proteasomes were found to be sequestered into IPODs when the proteasome functions were completely blocked by genetic aberration in yeast [107].
Interestingly, the proteasome-containing aggresomes formed by reversible MG132 inhibition in mammalian cells are under both autophagy clearance and proteasome reversion processes, suggesting that they are JUNQ-like fluidic foci, but not IPODs [106]. Given that PSGs function as cytoplasmic storage foci for normal proteasomes that can be fully reverted when carbon starvation is removed [87], these MG132-induced proteasome fluidic aggresomes may function as a temporary storage place for malfunctioning proteasomes awaiting recovery (Figure 3). The requirement of proteasomal ubiquitylation by STUB1 in the aggresomal formation of inhibited proteasomes may play a similar role of the free ubiquitin in PSGs, which are not only essential for the formation but also for keeping the fluidity. Given the presence of DUB activities by RPN11 and other DUB enzymes associated with proteasomes, a high concentration of free ubiquitin could be enriched in these foci. However, prolonged or more severed stresses, such as proteasome inhibition by irreversible inhibitors, epoxomicin and carfilzomib, or genetic aberrations, would completely block the activity of the holo-complex, thus changing the fluidity of the aggresome for converting into IPODs.
The presence of two fluidity statuses of mammalian proteasome-containing aggresomes, JUNQ and IPOD, implies that plant proteasome aggregates may have similar versions. For example, early plant growth inhibition by MG132 can be rapidly recovered upon being transferred to normal growth media. This may reflect the presence of JUNQ-like proteasome aggregates that are targeted by RPN10-medaited proteaphagy in MG132-treated seedlings [97] (Figure 3). Up to now, the researchers still do not know how plant proteasome aggregates are formed. Clearly, RPN10 is not essential, since proteasome-containing vacuolar aggregates were detected at least upon nitrogen starvation treatment in the RPN10-1 mutant [97]. However, in mammalian cells, the autophagy receptor, p62, is essential for both the formation of proteasome-containing aggresomes and the subsequent autophagy-lysosomal degradation [98][106]. Other proteins, such as the scaffold protein ALFY (autophagy-linked FYVE) and aggrephagy receptor NBR1, were discovered to be present in the protein aggregates thought to have a similar function to p62 for assisting aggregation and mediating autophagy degradation [105]. It is yet an unresolved question why both NBR1 and RPN10, the two hitherto plant autophagy receptors identified, are not essential to the formation of proteasome aggregates [97][108]. In yeast, Hsp42 chaperone proteins were found to be essential for the aggregation of genetically impaired proteasomes into IPODs [107]. Whether Hsp42 or yet unknown chaperone proteins are required for plant proteasome aggregates requires further investigation (Figure 3).
In addition to the presence of soluble and insoluble substances in cells, proteinaceous and membraneless subcellular structures are now considered as a third biophysical state of proteins assembled through LLPS. LLPS not only develops protein aggregates, but also forms many functional subcellular structures, such as centrosome, the nucleolus, Cajal bodies, and P-bodies [100]. Interestingly, p62 has been recently found to form proteolytically active nuclear condensates in mammalian cells that are generated through LLPS. Within these condensates, ubiquitylated substrates, 26S proteasomes, three enzymes involved in ubiquitylation, and DUBs are assembled along with p62 into a concentrated droplet for the efficient proteolysis of nuclear proteins and unassembled proteasomal subunits [109]. Therefore, active proteasome condensates, PSGs, JUNK-like proteasome aggregates, and proteasome IPODs, may represent LLPS-regulated biophysical dynamicity of proteasomes [93] (Figure 3).
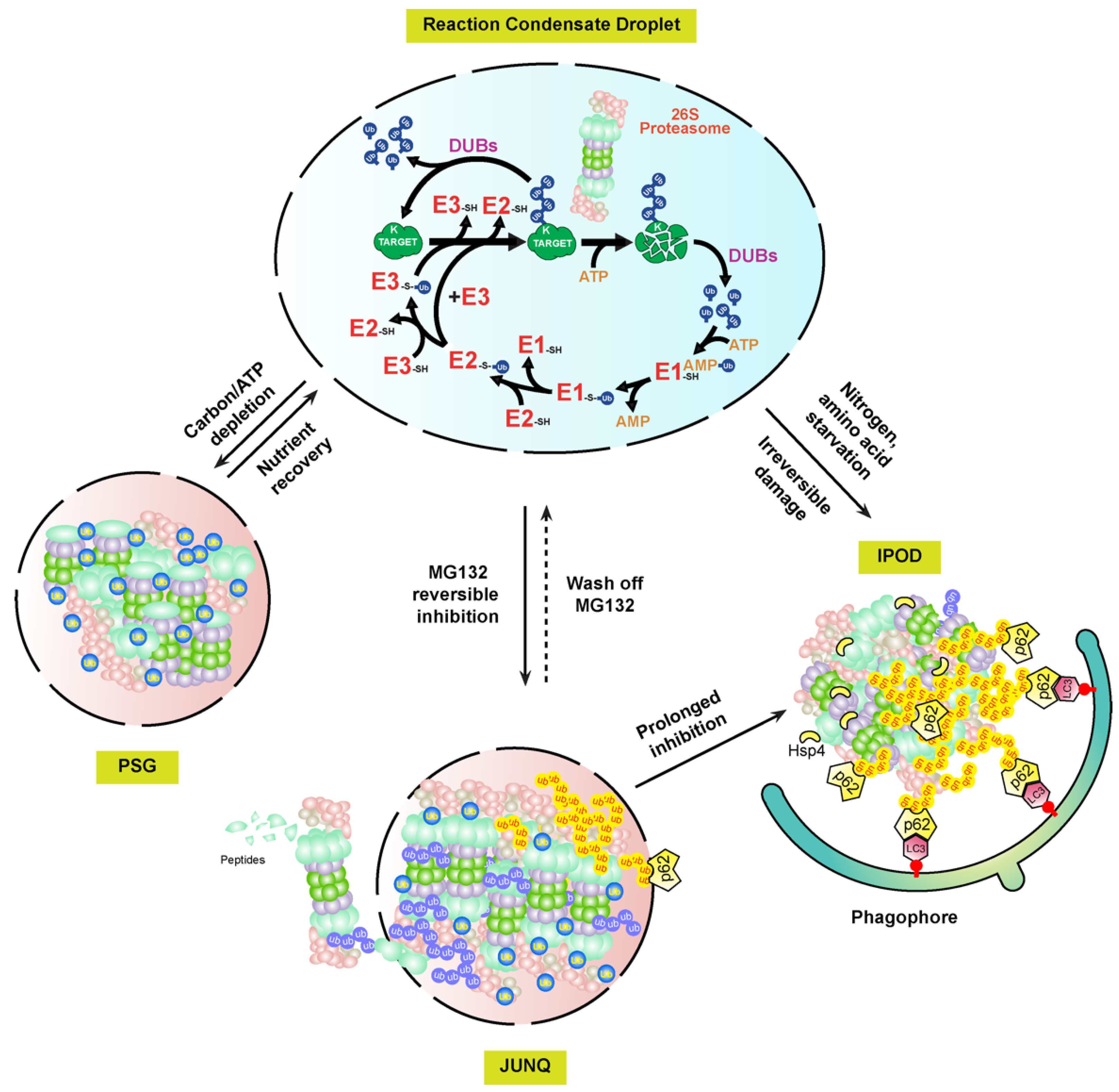
Figure 3. A biophysical dynamic module of intracellular proteasomes with different functional activities and stabilities. In normal cellular growth conditions, the UPS and its substrates may develop concentrated liquid droplets through LLPS for efficient proteolysis of ubiquitylation substrates [109]. Upon different stresses, the soluble proteasomes may either form PSG or JUNQ condensates, depending on whether the RP and CP subcomplexes are dissociated in yet unknown mechanisms [87][106]. The substances, including damaged proteasomes, in the highly fluidic JUNQ body are still under active ubiquitylation reactions. Ubiquitylated proteasome subunits may be extracted out of the JUNQ body for degradation by a functioning 26S proteasome, or directly targeted for autophagy-lysosomal/vacuolar degradation in either piecemeal or wholesale versions via autophagy receptors, e.g., p62 [110]. Continuing structure disruption by harsh or prolonged stresses may terminally sequester proteasome aggregates into IPOD foci, which are mediated by Hsp4 and only recognized by autophagy for selective degradation [107]. Proteasomes targeted by proteaphagy could be from either JUNQ or IPOD aggregates.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24032221
References
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471.
- Heinemeyer, W.; Fischer, M.; Krimmer, T.; Stachon, U.; Wolf, D.H. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 1997, 272, 25200–25209.
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403.
- Groll, M.; Bajorek, M.; Kohler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067.
- Choi, W.H.; de Poot, S.A.; Lee, J.H.; Kim, J.H.; Han, D.H.; Kim, Y.K.; Finley, D.; Lee, M.J. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun. 2016, 7, 10963.
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36.
- Hanna, J.; Finley, D. A proteasome for all occasions. FEBS Lett. 2007, 581, 2854–2861.
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55.
- Luan, B.; Huang, X.; Wu, J.; Mei, Z.; Wang, Y.; Xue, X.; Yan, C.; Wang, J.; Finley, D.J.; Shi, Y.; et al. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proc. Natl. Acad. Sci. USA 2016, 113, 2642–2647.
- Enenkel, C. Proteasome dynamics. Biochim. Biophys. Acta 2014, 1843, 39–46.
- Sahu, I.; Glickman, M.H. Proteasome in action: Substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021, 49, 629–644.
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513.
- Unverdorben, P.; Beck, F.; Sledz, P.; Schweitzer, A.; Pfeifer, G.; Plitzko, J.M.; Baumeister, W.; Forster, F. Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. USA 2014, 111, 5544–5549.
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191.
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, B.H.; Shi, Y.; Zhang, N.; de Poot, S.A.; Tuebing, F.; et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 2016, 351, 6275.
- VanderLinden, R.T.; Hemmis, C.W.; Yao, T.; Robinson, H.; Hill, C.P. Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem. 2017, 292, 9493–9504.
- Rosenzweig, R.; Bronner, V.; Zhang, D.; Fushman, D.; Glickman, M.H. Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. J. Biol. Chem. 2012, 287, 14659–14671.
- Deveraux, Q.; Ustrell, V.; Pickart, C.; Rechsteiner, M. A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 1994, 269, 7059–7061.
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488.
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477.
- Glickman, M.H.; Rubin, D.M.; Fried, V.A.; Finley, D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell Biol. 1998, 18, 3149–3162.
- Djuranovic, S.; Hartmann, M.D.; Habeck, M.; Ursinus, A.; Zwickl, P.; Martin, J.; Lupas, A.N.; Zeth, K. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol. Cell 2009, 34, 580–590.
- Tomko, R.J., Jr.; Funakoshi, M.; Schneider, K.; Wang, J.; Hochstrasser, M. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: Implications for proteasome structure and assembly. Mol. Cell 2010, 38, 393–403.
- Bard, J.A.M.; Bashore, C.; Dong, K.C.; Martin, A. The 26S proteasome utilizes a kinetic gateway to prioritize substrate degradation. Cell 2019, 177, 286–298.e15.
- Smith, D.M.; Chang, S.C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 2007, 27, 731–744.
- Erales, J.; Hoyt, M.A.; Troll, F.; Coffino, P. Functional asymmetries of proteasome translocase pore. J. Biol. Chem. 2012, 287, 18535–18543.
- Beckwith, R.; Estrin, E.; Worden, E.J.; Martin, A. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat. Struct. Mol. Biol. 2013, 20, 1164–1172.
- Prakash, S.; Tian, L.; Ratliff, K.S.; Lehotzky, R.E.; Matouschek, A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol. 2004, 11, 830–837.
- Fishbain, S.; Prakash, S.; Herrig, A.; Elsasser, S.; Matouschek, A. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat. Commun. 2011, 2, 192.
- Inobe, T.; Fishbain, S.; Prakash, S.; Matouschek, A. Defining the geometry of the two-component proteasome degron. Nat. Chem. Biol. 2011, 7, 161–167.
- Fishbain, S.; Inobe, T.; Israeli, E.; Chavali, S.; Yu, H.; Kago, G.; Babu, M.M.; Matouschek, A. Sequence composition of disordered regions fine-tunes protein half-life. Nat. Struct. Mol. Biol. 2015, 22, 214–221.
- Takeuchi, J.; Chen, H.; Coffino, P. Proteasome substrate degradation requires association plus extended peptide. EMBO J. 2007, 26, 123–131.
- Zhao, M.; Zhang, N.Y.; Zurawel, A.; Hansen, K.C.; Liu, C.W. Degradation of some polyubiquitinated proteins requires an intrinsic proteasomal binding element in the substrates. J. Biol. Chem. 2010, 285, 4771–4780.
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol. Cell 2010, 40, 671–681.
- Kusmierczyk, A.R.; Kunjappu, M.J.; Kim, R.Y.; Hochstrasser, M. A conserved 20S proteasome assembly factor requires a C-terminal HbYX motif for proteasomal precursor binding. Nat. Struct. Mol. Biol. 2011, 18, 622–629.
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.C.; Goldberg, A.L.; Cheng, Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell 2008, 30, 360–368.
- Chojnacki, M.; Mansour, W.; Hameed, D.S.; Singh, R.K.; El Oualid, F.; Rosenzweig, R.; Nakasone, M.A.; Yu, Z.; Glaser, F.; Kay, L.E.; et al. Polyubiquitin-photoactivatable crosslinking reagents for mapping ubiquitin interactome identify Rpn1 as a proteasome ubiquitin-associating subunit. Cell Chem. Biol. 2017, 24, 443–457.e6.
- Schweitzer, A.; Aufderheide, A.; Rudack, T.; Beck, F.; Pfeifer, G.; Plitzko, J.M.; Sakata, E.; Schulten, K.; Forster, F.; Baumeister, W. Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. USA 2016, 113, 7816–7821.
- Effantin, G.; Rosenzweig, R.; Glickman, M.H.; Steven, A.C. Electron microscopic evidence in support of alpha-solenoid models of proteasomal subunits Rpn1 and Rpn2. J. Mol. Biol. 2009, 386, 1204–1211.
- He, J.; Kulkarni, K.; da Fonseca, P.C.; Krutauz, D.; Glickman, M.H.; Barford, D.; Morris, E.P. The structure of the 26S proteasome subunit Rpn2 reveals its PC repeat domain as a closed toroid of two concentric alpha-helical rings. Structure 2012, 20, 513–521.
- Kajava, A.V. What curves alpha-solenoids? Evidence for an alpha-helical toroid structure of Rpn1 and Rpn2 proteins of the 26 S proteasome. J. Biol. Chem. 2002, 277, 49791–49798.
- Hamazaki, J.; Iemura, S.; Natsume, T.; Yashiroda, H.; Tanaka, K.; Murata, S. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J. 2006, 25, 4524–4536.
- Chen, X.; Lee, B.H.; Finley, D.; Walters, K.J. Structure of proteasome ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Mol. Cell 2010, 38, 404–415.
- Sakata, E.; Bohn, S.; Mihalache, O.; Kiss, P.; Beck, F.; Nagy, I.; Nickell, S.; Tanaka, K.; Saeki, Y.; Forster, F.; et al. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 1479–1484.
- Savulescu, A.F.; Rotem, A.; Harel, A. Proteasomes crossing the nuclear border. Nucleus 2011, 2, 258–263.
- Dambacher, C.M.; Worden, E.J.; Herzik, M.A.; Martin, A.; Lander, G.C. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. Elife 2016, 5, e13027.
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407.
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., 3rd; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615.
- Worden, E.J.; Padovani, C.; Martin, A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat. Struct. Mol. Biol. 2014, 21, 220–227.
- Pathare, G.R.; Nagy, I.; Sledz, P.; Anderson, D.J.; Zhou, H.J.; Pardon, E.; Steyaert, J.; Forster, F.; Bracher, A.; Baumeister, W. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc. Natl. Acad. Sci. USA 2014, 111, 2984–2989.
- Matyskiela, M.E.; Lander, G.C.; Martin, A. Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol. 2013, 20, 781–788.
- Ding, Z.; Fu, Z.; Xu, C.; Wang, Y.; Wang, Y.; Li, J.; Kong, L.; Chen, J.; Li, N.; Zhang, R.; et al. High-resolution cryo-EM structure of the proteasome in complex with ADP-AlFx. Cell Res. 2017, 27, 373–385.
- Sledz, P.; Unverdorben, P.; Beck, F.; Pfeifer, G.; Schweitzer, A.; Forster, F.; Baumeister, W. Structure of the 26S proteasome with ATP-gammaS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc. Natl. Acad. Sci. USA 2013, 110, 7264–7269.
- Wehmer, M.; Rudack, T.; Beck, F.; Aufderheide, A.; Pfeifer, G.; Plitzko, J.M.; Forster, F.; Schulten, K.; Baumeister, W.; Sakata, E. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. USA 2017, 114, 1305–1310.
- Eisele, M.R.; Reed, R.G.; Rudack, T.; Schweitzer, A.; Beck, F.; Nagy, I.; Pfeifer, G.; Plitzko, J.M.; Baumeister, W.; Tomko, R.J., Jr.; et al. Expanded coverage of the 26S proteasome conformational landscape reveals mechanisms of peptidase gating. Cell Rep. 2018, 24, 1301–1315.e5.
- de la Pena, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 2018, 362, eaav0725.
- Asano, S.; Fukuda, Y.; Beck, F.; Aufderheide, A.; Forster, F.; Danev, R.; Baumeister, W. Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 2015, 347, 439–442.
- Hua, Z.; Vierstra, R.D. The cullin-RING ubiquitin-protein ligases. Annu. Rev. Plant Biol. 2011, 62, 299–334.
- Linden, K.J.; Callis, J. The ubiquitin system affects agronomic plant traits. J. Biol. Chem. 2020, 295, 13940–13955.
- Copeland, C.; Li, X. Regulation of plant immunity by the proteasome. Int. Rev. Cell Mol. Biol. 2019, 343, 37–63.
- Doroodian, P.; Hua, Z. The ubiquitin switch in plant stress response. Plants 2021, 10, 246.
- Kelley, D.R. E3 ubiquitin ligases: Key regulators of hormone signaling in plants. Mol. Cell Proteom. 2018, 17, 1047–1054.
- Miricescu, A.; Goslin, K.; Graciet, E. Ubiquitylation in plants: Signaling hub for the integration of environmental signals. J. Exp. Bot. 2018, 69, 4511–4527.
- National Research Council (US) Committee on Examination of Plant Science Research Programs in the United States. Plant Biology Research and Training for the 21st Century; National Academies Press: Washington, DC, USA, 1992.
- Fu, H.; Doelling, J.H.; Arendt, C.S.; Hochstrasser, M.; Vierstra, R.D. Molecular organization of the 20S proteasome gene family from Arabidopsis thaliana. Genetics 1998, 149, 677–692.
- Fu, H.; Doelling, J.H.; Rubin, D.M.; Vierstra, R.D. Structural and functional analysis of the six regulatory particle triple-A ATPase subunits from the Arabidopsis 26S proteasome. Plant J. 1999, 18, 529–539.
- Fu, H.; Girod, P.A.; Doelling, J.H.; van Nocker, S.; Hochstrasser, M.; Finley, D.; Vierstra, R.D. Structure and functional analysis of the 26S proteasome subunits from plants. Mol. Biol. Rep. 1999, 26, 137–146.
- Hua, Z.; Yu, P. Diversifying evolution of the ubiquitin-26S proteasome system in Brassicaceae and Poaceae. Int. J. Mol. Sci. 2019, 20, 3226.
- Matsumoto, T.; Wu, J.; Itoh, T.; Numa, H.; Antonio, B.; Sasaki, T. The Nipponbare genome and the next-generation of rice genomics research in Japan. Rice 2016, 9, 33.
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313.
- Leggett, D.S.; Hanna, J.; Borodovsky, A.; Crosas, B.; Schmidt, M.; Baker, R.T.; Walz, T.; Ploegh, H.; Finley, D. Multiple associated proteins regulate proteasome structure and function. Mol. Cell 2002, 10, 495–507.
- Book, A.J.; Gladman, N.P.; Lee, S.S.; Scalf, M.; Smith, L.M.; Vierstra, R.D. Affinity purification of the Arabidopsis 26 S proteasome reveals a diverse array of plant proteolytic complexes. J. Biol. Chem. 2010, 285, 25554–25569.
- Dahlmann, B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch Biochem. Biophys. 2016, 591, 132–140.
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931.
- Yu, P.; Hua, Z. The ubiquitin-26S proteasome system and autophagy relay proteome homeostasis regulation during silique development. Plant J. 2022, 111, 1324–1339.
- Gemperline, D.C.; Marshall, R.S.; Lee, K.H.; Zhao, Q.; Hu, W.; McLoughlin, F.; Scalf, M.; Smith, L.M.; Vierstra, R.D. Proteomic analysis of affinity-purified 26S proteasomes identifies a suite of assembly chaperones in Arabidopsis. J. Biol. Chem. 2019, 294, 17570–17592.
- Marshall, R.S.; Vierstra, R.D. Dynamic regulation of the 26S proteasome: From synthesis to degradation. Front. Mol. Biosci. 2019, 6, 40.
- Yedidi, R.S.; Fatehi, A.K.; Enenkel, C. Proteasome dynamics between proliferation and quiescence stages of Saccharomyces cerevisiae. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 497–512.
- Mao, Y. Structure, dynamics and function of the 26S proteasome. Subcell. Biochem. 2021, 96, 1–151.
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome structure and assembly. J. Mol. Biol. 2017, 429, 3500–3524.
- Kato, K.; Satoh, T. Structural insights on the dynamics of proteasome formation. Biophys. Rev. 2018, 10, 597–604.
- Karmon, O.; Ben Aroya, S. Spatial organization of proteasome aggregates in the regulation of proteasome homeostasis. Front. Mol. Biosci. 2019, 6, 150.
- Enenkel, C.; Lehmann, A.; Kloetzel, P.M. GFP-labelling of 26S proteasomes in living yeast: Insight into proteasomal functions at the nuclear envelope/rough ER. Mol. Biol. Rep. 1999, 26, 131–135.
- Russell, S.J.; Steger, K.A.; Johnston, S.A. Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. J. Biol. Chem. 1999, 274, 21943–21952.
- Laporte, D.; Lebaudy, A.; Sahin, A.; Pinson, B.; Ceschin, J.; Daignan-Fornier, B.; Sagot, I. Metabolic status rather than cell cycle signals control quiescence entry and exit. J. Cell Biol. 2011, 192, 949–957.
- Narayanaswamy, R.; Levy, M.; Tsechansky, M.; Stovall, G.M.; O’Connell, J.D.; Mirrielees, J.; Ellington, A.D.; Marcotte, E.M. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. USA 2009, 106, 10147–10152.
- Laporte, D.; Salin, B.; Daignan-Fornier, B.; Sagot, I. Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. J. Cell Biol. 2008, 181, 737–745.
- Saunier, R.; Esposito, M.; Dassa, E.P.; Delahodde, A. Integrity of the Saccharomyces cerevisiae Rpn11 protein is critical for formation of proteasome storage granules (PSG) and survival in stationary phase. PLoS ONE 2013, 8, e70357.
- Peters, L.Z.; Karmon, O.; Miodownik, S.; Ben-Aroya, S. Proteasome storage granules are transiently associated with the insoluble protein deposit in Saccharomyces cerevisiae. J. Cell Sci. 2016, 129, 1190–1197.
- Bajorek, M.; Finley, D.; Glickman, M.H. Proteasome disassembly and downregulation is correlated with viability during stationary phase. Curr. Biol. 2003, 13, 1140–1144.
- Weberruss, M.H.; Savulescu, A.F.; Jando, J.; Bissinger, T.; Harel, A.; Glickman, M.H.; Enenkel, C. Blm10 facilitates nuclear import of proteasome core particles. EMBO J. 2013, 32, 2697–2707.
- Gu, Z.C.; Wu, E.; Sailer, C.; Jando, J.; Styles, E.; Eisenkolb, I.; Kuschel, M.; Bitschar, K.; Wang, X.; Huang, L.; et al. Ubiquitin orchestrates proteasome dynamics between proliferation and quiescence in yeast. Mol. Biol. Cell 2017, 28, 2479–2491.
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434.
- Aufderheide, A.; Unverdorben, P.; Baumeister, W.; Forster, F. Structural disorder and its role in proteasomal degradation. FEBS Lett. 2015, 589 Pt A, 2552–2560.
- Enenkel, C. The paradox of proteasome granules. Curr. Genet. 2018, 64, 137–140.
- Kolodziejek, I.; Misas-Villamil, J.C.; Kaschani, F.; Clerc, J.; Gu, C.; Krahn, D.; Niessen, S.; Verdoes, M.; Willems, L.I.; Overkleeft, H.S.; et al. Proteasome activity imaging and profiling characterizes bacterial effector syringolin A. Plant Physiol. 2011, 155, 477–489.
- Marshall, R.S.; Li, F.; Gemperline, D.C.; Book, A.J.; Vierstra, R.D. Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis. Mol. Cell 2015, 58, 1053–1066.
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499.
- Waite, K.A.; Burris, A.; Vontz, G.; Lang, A.; Roelofs, J. Proteaphagy is specifically regulated and requires factors dispensable for general autophagy. J. Biol. Chem. 2022, 298, 101494.
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822.
- Ustun, S.; Hafren, A.; Liu, Q.; Marshall, R.S.; Minina, E.A.; Bozhkov, P.V.; Vierstra, R.D.; Hofius, D. Bacteria exploit autophagy for proteasome degradation and enhanced virulence in plants. Plant Cell 2018, 30, 668–685.
- Waite, K.A.; De-La Mota-Peynado, A.; Vontz, G.; Roelofs, J. Starvation induces proteasome autophagy with different pathways for core and regulatory particles. J. Biol. Chem. 2016, 291, 3239–3253.
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530.
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095.
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905.
- Choi, W.H.; Yun, Y.; Park, S.; Jeon, J.H.; Lee, J.; Lee, J.H.; Yang, S.A.; Kim, N.K.; Jung, C.H.; Kwon, Y.T.; et al. Aggresomal sequestration and STUB1-mediated ubiquitylation during mammalian proteaphagy of inhibited proteasomes. Proc. Natl. Acad. Sci. USA 2020, 117, 19190–19200.
- Peters, L.Z.; Karmon, O.; David-Kadoch, G.; Hazan, R.; Yu, T.; Glickman, M.H.; Ben-Aroya, S. The protein quality control machinery regulates its misassembled proteasome subunits. PLoS Genet. 2015, 11, e1005178.
- Jung, H.; Lee, H.N.; Marshall, R.S.; Lomax, A.W.; Yoon, M.J.; Kim, J.; Kim, J.H.; Vierstra, R.D.; Chung, T. Arabidopsis cargo receptor NBR1 mediates selective autophagy of defective proteins. J. Exp. Bot. 2020, 71, 73–89.
- Fu, A.; Cohen-Kaplan, V.; Avni, N.; Livneh, I.; Ciechanover, A. p62-containing, proteolytically active nuclear condensates, increase the efficiency of the ubiquitin-proteasome system. Proc. Natl. Acad. Sci. USA 2021, 118, e2107321118.
- Clavel, M.; Dagdas, Y. Proteasome and selective autophagy: Brothers-in-arms for organelle quality control. Curr. Opin. Plant Biol. 2021, 63, 102106.
This entry is offline, you can click here to edit this entry!