Despite tremendous progresses in therapy in the last few years, cardiovascular diseases are still the leading cause of death worldwide [
1,
2]. To date, 18 million people die each year due to cardiovascular diseases [
1,
3,
4,
5,
6,
7]. These numbers are expected to increase, reaching 24 million yearly deaths worldwide from cardiovascular-related disease by 2030, with an average of over 66,000 per day, and a total global cost rising to over 1 trillion USD [
1,
3,
4,
5,
6,
7]. After a decline in mortality in recent decades, the numbers are rising again, reversing years of progress in both ischemic heart and cerebrovascular disease [
1,
4]. Thus, cardiovascular prevention is essential to make a 25% reduction in premature mortality from non-communicable diseases more realistic. There is an urgent need for plans and policies to reduce the burden of cardiovascular diseases which are a health challenge, starting from the territory and the role of cities in promoting health. There is also a need for improving the understanding of the basic mechanisms underlying cardiovascular diseases to design tailored therapy.
The atherosclerotic process is very often responsible for several cardiovascular and cerebrovascular diseases [
8]. It is now well accepted and documented that atherosclerosis starts from endothelial dysfunction and lipid deposition, which progresses through macrophage infiltration, smooth muscle cell migration, and blood borne material deposition and becomes clinically relevant due to complications, eventually leading to local intravascular thrombus formation [
8,
9]. Modified lipoproteins, mainly oxidized low-density lipoproteins (oxLDL), are considered the major contributors to the genesis, progression, and immunological response occurring during the atherosclerotic process [
10,
11]. The first report linking mitochondria to atherosclerosis is from 1970 [
12]. However, only in the last few years has increasing evidence really underlined the key role of mitochondrial dynamics in the pathogenesis of atherosclerosis [
13]. Vascular cells, such as endothelial and smooth muscle cells, due to their metabolic functions and their barrier role are the main targets of mitochondrial dysfunction. In the atherosclerosis process, dysfunctional mitochondria might cause alterations in cellular metabolism and respiration resulting in the excessive production of reactive oxygen species (ROS), leading to oxidative stress [
13]. While low levels of ROS exert important signaling functions [
14,
15], elevated ROS production induces the damage of cellular structures, alters DNA, proteins, and other molecules [
16]. These conditions can become chronic, thereby favoring atherosclerosis progression and destabilization [
17]. It is important to point out that mitochondrial dysfunction can be inherited and acquired [
18]. Primary mitochondrial disease (PMD) is clinically diagnosed and confirmed by pathogenic mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) mutations [
18]. However, there are some disorders with a ‘mitochondrial’ phenotype without identifiable mtDNA or nDNA mutations, or with variants of unknown clinical significance [
18]. Conversely, secondary mitochondrial dysfunction (SMD) can be caused by genes encoding neither function nor production of oxphos proteins, and associated with many hereditary non-mitochondrial diseases [
18]. This SMD can also be related to nongenetic causes, such as environmental factors and ageing. The strong relationship between ageing, atherosclerosis, and cardiovascular diseases is well established [
19], and a correlation between mitochondrial dysfunction–ageing, and vice-versa, cannot be ruled out [
20].
2. Mechanism of Mitochondrial Dysfunction and Molecular Implication for the Cardiovascular System
Mitochondria represent the engine of human cells generating ATP, and their function is regulated via mitophagy. However, mitochondria are also involved in the production of metabolites for protein assembling and signal transduction. Their metabolism is influenced by Ca
2+ levels: low calcium concentrations can lead to mitochondrial dysfunction, while high levels of calcium can increase mitochondrial permeability [
21].
As compared to other organs, the heart needs a high amount of energy; this is produced by oxidative metabolism in the mitochondria, which are one-third of the total volume of cardiomyocytes [
22].
Mitochondrial dysfunctions are strongly related to cardiovascular diseases, in particular ischemic heart disease and atherosclerosis, cardiomyopathy, and hypertension [
23]. This altered mitochondrial function results in reduced ATP, impairment of mitochondrial regulatory roles, reactive oxygen species (ROS) production and related signaling, cell growth and apoptosis, impaired mitochondrial electron transport chain activity, and an inflammatory response [
24,
25,
26,
27].
Mitochondrial fission and fusion impairments have been related to the development of cardiovascular diseases [
28]. Novel mitochondrial biomarkers have been proposed to identify patients that can benefit from therapies specifically targeting mitochondria [
29].
Mitochondrial homeostasis plays a crucial role in cells with high energy consumption, and in the pathological development of ischemic heart disease [
30]. In particular, variation in the level of Ca
2+ in mitochondria could lead to altered contraction coupling [
31] and increased ROS levels during ischemic myocardial reperfusion, thereby inducing mitochondrial membrane damage, abnormal ATP synthesis, increased levels of Ca
2+, and mPTP opening [
32].
Thus, during ischemia, the mitochondrial succinate significantly increases in injured tissue [
33,
34], while during reperfusion, the accumulated metabolites are oxidized by the mitochondrial respiratory chain enzyme SDH (succinate dehydrogenase). This drives reactive oxygen species (ROS) production by reverse electron transport (RET) at mitochondrial complex I, stimulating mitochondrial permeability transition pore opening, and cell death associated with ischemia/reperfusion damage [
35,
36,
37,
38]. Sazanova et al. have demonstrated that specific mutations in the coding region of the mitochondrial genome, in particular m.14459G>A and m.5178C>A, are risk factors for occurrence and development of cardiac angina, while the mutation m.15059G>A instead had a protective effect [
39].
Atherosclerosis is a chronic inflammatory condition characterized by impaired lipid metabolism, which stimulates innate and adaptive immune responses [
9]. The presence of lipids in the intimal layer of arteries significantly affects mitochondrial activity/function. There are several mechanisms related to atherosclerosis progression and plaque instability, such as mutations in mitochondrial DNA (mtDNA), reactive oxidative species (ROS), and respiratory chain alterations, which induce hypertrophy of vascular smooth myocytes [
40]. Mitochondrial ROS, associated with several risk factors, such as hyperglycemia and hypercholesterolemia [
41], are involved in the expansion and potential damage of atherosclerotic plaques, particularly by inducing endothelial dysfunction, promoting monocyte infiltration, and increasing vascular smooth muscle cell and endothelial cell apoptosis [
42].
Accumulation of ROS in the inner mitochondrial membrane can alter the mitochondrial cholesterol transporter, steroidogenic acute regulatory protein (StAR), which inhibits the storage of cholesterol through the mitochondrial membrane [
43].
Under pathophysiological conditions, the dysfunctional mitochondria can stimulate the production of large amounts of ROS from adjacent mitochondria, modifying membrane potential, a process well known as “ROS-induced ROS”. Normally, this process of ROS is balanced by endogenous antioxidants. On the other hand, when there is an oxidative stress and hyper-production of ROS, the oxidative impairment in the arterial wall is increased [
44], and a progression of the atherosclerotic plaque can be activated [
45].
Mutations in mtDNA, related to ROS derived from mitochondrial dysfunction, are more frequent than in nuclear DNA and can cause impaired mitochondrial respiration in smooth muscle monocytes and macrophages, thereby contributing to atherosclerotic plaque progression [
41]. In vitro studies have demonstrated that the damage induced byROS on mtDNA inhibits protein synthesis and alters gene expression, suggesting that ROS might mediate vascular cell dysfunction in the setting of atherogenesis [
46] associated with changes in mitochondrial genome [
47].
On the other hand, recent studies have put into question the correlation between ROS and mtDNA damage, highlighting that impaired mtDNA was observed in vascular and circulating cells before the development of atherosclerotic lesions [
46]. Thus, these data suggested that the primary event is associated with mtDNA damage which increases ROS generation and variations in mitochondrial dysfunction and membrane potential, followed by activation of the apoptotic mechanism. Moreover, endogenous mitochondrial damage-associated molecular patterns (mtDAMPs) trigger sterile inflammation through various signaling pathways, including toll-like receptors (TLRs), nuclear factor kappa beta (NF-κβ), and the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome [
48]. In addition, damaged mtDNA can be seen as an endogenous damage-associated molecular pattern (DAMP), activating the inflammatory response [
49].
3. Mitochondrial Dysfunction and Endothelial Dysfunction
Endothelial cells represent the barrier for the molecular transport between blood and tissue, regulating the exchange of nutrients [
53] and the vascular tone by production of nitric oxide (NO) and contractile factors. Nevertheless, in addition to its function as a barrier, the endothelium modulates inflammation, lipid metabolism, vascular tone, and hemostasis, and prevents penetration of inflammatory cells into the bloodstream. It also secretes adhesion molecules and cytokines involved in the inflammatory cascade [
54,
55].
Endothelial cells’ dysfunction is characterized by ROS production, in particular from mitochondria, infiltrations of LDL, and subsequent oxidation to oxLDL [
56], which might induce endothelial impairment. This enhances the expression of adhesion molecules ICAM-1, VCAM-1, and P-selectins, resulting in smooth muscle cell growth and activation of inflammatory cells, such as monocytes and macrophages, thereby causing a pro-inflammatory burden by releasing cytokines and increasing circulating immune cells’ adhesion to the endothelium [
57].
Damaged endothelium produces growth factors, which activates smooth muscle cells (SMCs) in the vascular bed and the phagocytosis of lipids, with subsequent evolution into the fibrous cap [
58].
Mitochondria are important regulators of apoptosis and NO production in vascular endothelial cells, stimulating the cellular response to stress [
59]. At the same time, the amounts of mitochondria in the endothelium are not abundant, but despite that, mitochondria not only affect energy supply, but also regulate blood oxygen levels and mediate NO-mediated vasodilation [
60,
61]. The decreased NO production in atherosclerotic plaques is mainly due to endothelial NO synthase (eNOS) degradation, induced by ROS-mediated oxidative stress. In addition, eNOS decoupling dysfunction produces additional ROS, which interfere with endothelial function, inducing the progression of atherosclerosis [
62].
Recent studies have highlighted that the transcription factor NF-E2 p45-related factor 2 (Nrf2; gene name
NFE2L2) regulates some mitochondrial functions [
63] via the mtROS pathway, such as antioxidant activity, autophagy, and metabolism [
64]. Nrf2 has a crucial role in the maintenance of cellular redox homeostasis by regulating biosynthesis, utilization, and regeneration of glutathione, thioredoxin, and NADPH, and controlling the production of ROS by mitochondria and NADPH oxidase. Moreover, Nrf2 activation inhibits Drp1-mediated mitochondrial fission, improving endothelial dysfunction [
65].
Incubation of endothelial cells with oxLDL leads to an increase in the activity of mitochondrial complex I and oxidative stress [
66], thereby stimulating the transcription and expression of superoxide dismutase 2 (SOD2) in macrophages [
67].
Activation of mitophagy, a lysosome-mediated selective mitochondria degradation [
68], is one of the characteristic signs of mitochondrial dysfunction and damage and leads to membrane potential collapse, increased ROS production, and decreased ATP levels, oxidative stress, and apoptosis [
69]. The balance between mitophagy and mitochondrial neogenesis is crucial for correct homeostasis in the human body [
70]. Mitochondrial biogenesis is an important process for maintaining energy control and protecting endothelial cells’ survival in critical pathological scenarios [
71,
72]. Mitochondrial dysfunction might represent the first real step of atherosclerosis by determining the endothelial impairment, which is the starting point of the atherosclerotic process [
21].
The endothelial dysfunction, in particular decreased vasodilatation, is also typical of MELAS (Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) patients [
73].
These patients can have lower levels of l-arginine, which regulates the endothelial-dependent vascular relaxation [
74], which was significantly lowered in both acute and interictal phases of MELAS as compared to control subjects [
75].
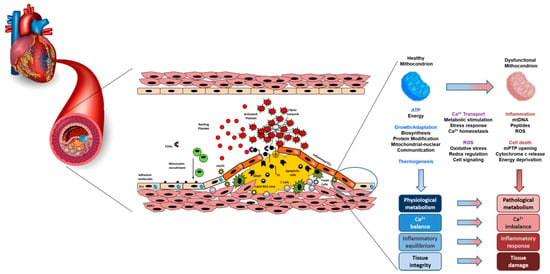
A schematic view of the atherosclerotic mitochondria-related process is shown in Figure 1.
Figure 1. A schematic view of the interaction between mitochondria dysfunction, atherosclerosis and oxidative stress.