Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Abscisic acid (ABA), long known as a plant stress hormone, is present and functionally active in organisms other than those pertaining to the land plant kingdom, including cyanobacteria, fungi, algae, protozoan parasites, lower Metazoa, and mammals.
- AMPK
- PGC-1α
- Sirt1/3
- LANCL1/2
- mitochondrial proton gradient
1. The ABA/LANCL1/2 Hormone/Receptor System
1.1. Abscisic Acid, an Ancient Stress Signal
Abscisic acid is an ancient signal molecule, which likely evolved in primordial unicellular organisms, much earlier than the divergence of plants and animals. It is present and active in modern bacteria, unicellular algae, basal and vascular plants, as well as mammals, including humans. In the more complex Metaphyta and Metazoa, ABA has a conserved role as a stress hormone, adapting cell, tissue, and organism functions to environmental stimuli; it is perhaps the oldest cross-kingdom hormone known to date.
Although the molecule itself is simple compared with other hormones (an isoprenoid structure with a mol. weight of 264, Figure 1), the perception and signaling pathways of ABA are quite complex and distinct in divergent organisms, such as plants and mammals. At the functional level, however, strikingly conserved effects of ABA can be observed in organisms as evolutionarily distant as land plants and mammals.
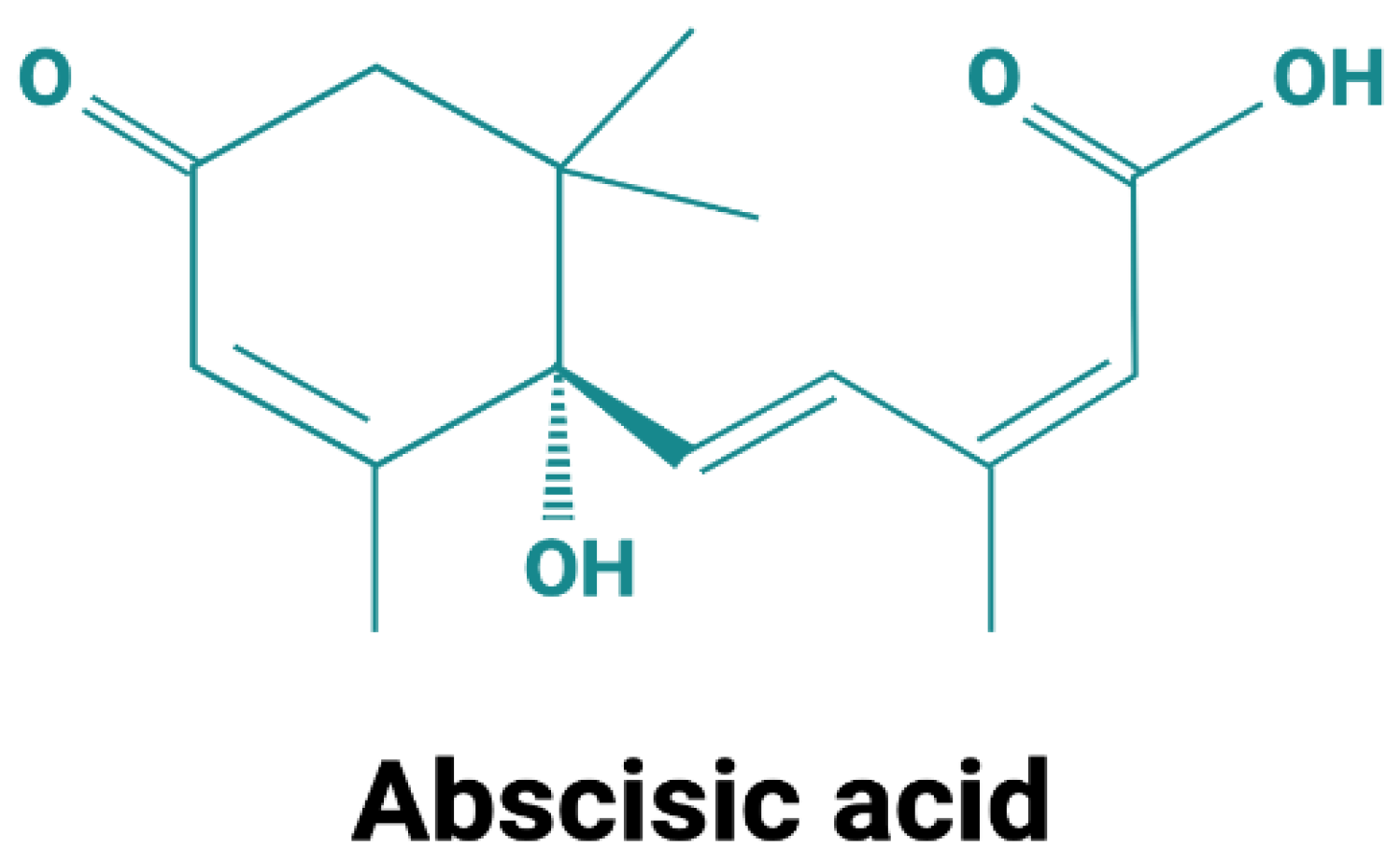
Figure 1. Structure of abscisic acid. Abscisic acid exists in two enantiomers: S-(+)-ABA, which is the predominant form in plants, and R-(−)-ABA. Most studies in mammals employed a racemic mixture. When individually tested on innate immune cells, both ABA enantiomers were similarly effective in the induction of an intracellular Ca2+ increase and in stimulating chemotaxis [1].
For instance, ABA leads to nitric oxide (NO)-mediated stomatal closure in response to several stressors, including UV light, in plants [2], and human keratinocytes exposed to UV light release ABA, which stimulates NO production by these cells [3]. In human innate immune cells, physical or chemical stimuli induce ABA release and ABA stimulates NO production among other functional responses, such as migration and phagocytosis [3]. It could be argued that NO may be another of those few molecular signals that evolved very early in primordial cells. This ABA/NO-mediated “alarm pathway” in response to an environmental stress is indeed conserved across kingdoms [4].
The quest for plant and animal ABA receptors has been underway for decades and is still a topic of intense investigation; recent evolutionary studies indicate that ABA was present before its (modern) specific receptor PYL acquired the ability to be hormonally modulated by it, as occurs in land plants [5].
1.2. Mammalian LANCL Proteins
LANCL (Lan C-like) proteins derive their name from their sequence homology with bacterial lanthionine synthetase, which synthesizes lantibiotics, a class of natural antibiotics originating from modified cysteine. The mammalian LANCL family comprises three proteins: LANCL1 is the most highly expressed, particularly in the brain, and is a cytosolic protein also loosely associated with the plasmamembrane [6]. LANCL2 is membrane-anchored through its myristoylated N-terminal [7], and LANCL3 has the lowest expression levels of the LANCL proteins and may be a pseudogene. The three LANCL proteins share a significant (approx. 35%) sequence identity, likely arising from gene duplication. As the triple knockdown in mice does not reduce the brain content of a downstream metabolite of lanthionine, lanthionine ketimine, it has been concluded that mammalian LANCL proteins are not involved in lanthionine synthesis [8]. Interestingly, however, triple LANCL knock-out (KO) mice die prematurely [9], indicating an important role for LANCL proteins in animal physiology, which may be linked to the features described in this research.
LANCL proteins also show a significant (approx. 30%) sequence identity with another evolutionary distant protein, the plant orphan, G-protein linked receptor GCR2, which was proposed as an ABA receptor in Arabidopsis [10], a discovery subsequently obscured, but not dismissed [11][12], by the discovery of the PYR/PYL/RCAR family of ABA-sensing proteins in higher plants [5].
Still, the sequence homology between plant GPCR-type ABA receptor and mammalian LANCL proteins spurred research on their possible role as mammalian ABA receptors.
1.2.1. The Membrane-Bound LANCL2 Receptor
The LANCL2 protein first attracted interest as a putative mammalian ABA receptor; indeed, in vitro studies using several different techniques demonstrate high-affinity ABA binding to human recombinant LANCL2 (Kd 2.6 nM) [13], and LANCL2 is required for ABA action in several mammalian cell types [14]. LANCL2 has an unusual behavior as a hormone receptor: it is coupled to a G protein when membrane bound, but it is de-myristoylated upon ABA binding, detaching from the membrane and accumulating in the cell nucleus [15]. This behavior combines features typical of the receptors for peptide (G protein coupling) and for steroid hormones (nuclear translocation), perhaps a heritage of the primordial origin of the hormone, when Nature was still experimenting with its signaling pathways, or the result of the hydrophilic or lipophilic nature of dissociated or of protonated ABA, respectively. The binding of ABA to LANCL2 on the inner plasma membrane layer requires influx of the hormone through the plasma membrane, which occurs through transporters of the anion exchanger (AE) superfamily [16]. Protonated ABA can instead diffuse through the lipid bilayer; however, a very low percentage of ABA is protonated at the near-neutral pH of plasma and interstitial fluids. Thus, the exchange of ABA between extra- and intracellular fluids requires a transport system. An area where ABA transport is not needed to cross barriers is the gastric environment, where the strongly acidic pH favors the protonated, membrane-permeant form of the molecule, allowing diffusion of ABA across the gastric lipid bilayer. This fact probably accounts for the rapid absorption of the hormone after oral intake [17].
1.2.2. LANCL1 Is Also an ABA Receptor
A reduction, though not the complete abrogation, of the effect of ABA occurs in adipose and muscle cells after LANCL2 silencing [18][19], suggesting a role for other receptors in the metabolic action of the hormone. A more direct indication that other receptors could contribute to mediate the metabolic actions of ABA comes from studies on LANCL2 KO mice. LANCL2 KO mice show a reduced glucose tolerance compared with wild-type siblings; however, they are still responsive to exogenous ABA (1 μg/kg body weight (BW)/day), which significantly reduces the AUC of glycemia after glucose load to values similar to those of wild-type animals [19]. On the one hand, these results indicate that the absence of LANCL2 impairs glucose tolerance; on the other hand, they indicate that another receptor can substitute for LANCL2 in the stimulation of muscle and adipose tissue glucose transport, although at higher ABA concentrations than those reached by the endogenous hormone. Indeed, upon intake of ABA at a dose of 1 μg/kg BW plasma, ABA (ABAp) increases between 10 and 50 times compared with endogenous levels in humans [17]. The increase in ABAp achieved by exogenous ABA supplementation can apparently recruit a receptor with lower affinity, which may not be activated by lower endogenous ABA levels. An obvious candidate for this role is LANCL1, as it shares a significant sequence identity (54.2%), similar intracellular localization (it is membrane-associated, although not membrane-bound), and tissue expression pattern with LANCL2.
Interestingly, silencing or genetic ablation of LANCL2 in cells, or in mice, results in the spontaneous overexpression of LANCL1. Similarly, silencing of LANCL1 results in a significant increase in the expression of LANCL2 in L6 muscle cells [19]. These observations, together with the redundancy of ABA receptors, point to the physiological relevance of the ABA/LANCL hormone/receptor system in mammals. From a functional perspective, LANCL1 binds ABA with a somewhat lower affinity compared with LANCL2, but it activates the same signaling pathway (the AMPK/PGC-1α/Sirt1 axis), resulting in similar transcriptional and functional responses (increased glucose uptake and metabolism, mitochondrial respiration, and uncoupling), in vitro in rat myoblasts and in vivo in the skeletal muscle of LANCL2 KO mice [19].
1.2.3. AMPK Activation Downstream of LANCL1/2
Experiments performed in vitro on L6 myoblasts and ex vivo on murine skeletal muscle demonstrate that ABA stimulates muscle glucose transport in the absence of insulin and that activation of AMPK is responsible for this effect, as it is abrogated by the inhibition of AMPK with dorsomorphin. ABA indeed increases the phosphorylation of AMPK on Thr172 in L6 cells and in mouse muscle and also stimulates AMPK transcription [20]. Preincubation of L6 myoblasts with AZD5363, at pan-Akt competitive inhibitor, significantly increased pAMPK levels in ABA-treated compared with untreated cells, indicating the presence of an inhibitory effect by Akt on ABA-induced AMPK activation [20]. Indeed, the activation of Akt by phosphorylation on both Thr308 and Ser473 inhibits AMPK phosphorylation on Thr172 by LKB1 [21][22]. Akt lies at the crossroads between the starved and fed state: in the fed state, insulin favors double phosphorylation (on Ser473 and on Thr308), and maximal activation of Akt [22], which triggers cell energy storage, mainly via glycogen and triglyceride synthesis, concomitantly inhibiting AMPK phosphorylation and activation by LKB1. Conversely, AMPK activation occurs under conditions of reduced cell energy availability and stimulates metabolic and mitochondrial functions aimed at restoring energy balance. Stimulation of cell glucose uptake is a common feature of both ABA- (via AMPK) and insulin- (via Akt) triggered signaling, enabling energy production and energy storage, respectively. Glucose uptake, detected in skeletal muscle by dynamic micro-PET, increases 2-fold in ABA-treated compared with untreated rats during an oral glucose load [20]. Given the high percentage of BW represented by skeletal muscle in rodents, approximately 45%, the increased muscle glucose uptake is likely responsible for the accelerated blood glucose clearance observed in ABA-treated compared with control mice.
1.2.4. ABA Signaling in Skeletal Muscle
The signaling pathway activated by ABA in the skeletal muscle involves the AMPK/PGC-1α/Sirt1 axis, resulting in the increased gene transcription and protein overexpression of the glucose transporters GLUT1 and GLUT4, of the NAD-synthesizing enzyme Nampt, of RabGAP TBC1D1, and of the muscle-specific mitochondrial uncoupling proteins UCP-3 and sarcolipin and in an increased mitochondrial DNA content [19]. These transcriptional and translational effects of ABA increase muscle glucose uptake and energy metabolism, leading to increased muscle glucose consumption. The increased muscle expression of LANCL1 in LANCL2 KO mice as compared with wild-type siblings may explain why LANCL2 KO mice with streptozotocin (STZ)-induced diabetes indeed respond to ABA similarly to, or perhaps even better than, wild-type mice. In addition, LANCL2 KO mice have a higher skeletal muscle mitochondrial DNA content and increased expression levels of AMPK, PGC-1α, GLUT1/4, Nampt, and UCP-3 compared with wild-type mice, levels which further increase after chronic ABA administration. Researchers observed a significant increase in the transcription of key glycolytic enzymes (GaPDH, PFK1) and of PDH in the skeletal muscle of ABA-treated wild-type and LANCL2 KO mice as compared with untreated controls, which is expected to stimulate oxidative muscle glucose consumption. Interestingly, LANCL1-overexpressing LANCL2 KO female mice fed a high-glucose diet for three months had a significantly lower body weight gain as compared with wild-type siblings under higher food intake. These findings suggest that muscle and adipocyte mitochondrial uncoupling and increased oxygen consumption controlled by the ABA/LANCL system may affect whole body energy consumption. At the end point of a similar protocol of ABA-pretreatment followed by diabetes induction with low-dose STZ, the mean glycemia of LANCL2 KO mice was significantly lower than that of wild-type animals. These effects can be attributed to the overexpression of LANCL1, which can substitute for LANCL2 in binding ABA and activating its signaling pathway.
Finally, the increased transcription of the insulin receptor mRNA in the skeletal muscle from chronically ABA-treated mice suggests that long-term treatment with ABA may improve muscle sensitivity to both endogenous and exogenous insulin. Probably as a consequence of the increased glucose uptake and oxidation, a markedly increased physical performance on a running wheel was observed in ABA-treated mice compared with untreated controls [20].
1.2.5. The ABA/LANCL System in the Adipose Tissue
Adipose tissue (AT) is one of the largest organs in the body and plays an important role in energy balance and glucose and lipid homeostasis. In mammals, white adipose tissue (WAT), diffused subcutaneously and abdominally, is specialized in energy (triglyceride) storage; conversely, brown adipose tissue (BAT), which is much less abundant than WAT and has specific localization sites (cervical, supraclavicular, paravertebral, and supra-adrenal in adult humans), is specialized in energy expenditure. Excess visceral WAT is currently believed to be conducive to the metabolic syndrome, overt diabetes, and cardiovascular disease due to a state of low-grade inflammation originating in, and maintained by, adipokines produced in WAT and cytokines produced by WAT-infiltrating inflammatory cells. Thus, the observation that ABA can reduce WAT inflammation in mice fed a high-fat diet [23] is particularly important in view of the possible use of ABA-containing nutraceuticals to combat low-grade inflammation in the adipose tissue. Pointedly, high-sensitivity C-reactive protein levels (hs-CRP, a measure of chronic, low-grade inflammation) were significantly reduced in prediabetic subjects after treatment with an ABA-rich vegetal extract compared with untreated controls [24]. Thermogenesis by BAT (the so-called non-shivering thermogenesis) is activated by cold stress and is controlled by sympathetic innervation and by several hormonal signals (T3, adenosine): heat production occurs due to the partial uncoupling of mitochondrial oxidative phosphorylation during the oxidation of glucose and of triglyceride-derived fatty acids. These features of BAT make it an attractive target for therapeutic strategies aimed at stimulating BAT activity to reduce body weight. Alas, at variance with WAT, BAT only accounts for a minute percentage of total AT; thus, another currently pursued strategy to increase whole-body energy expenditure is to induce the development of brown fat-like cells (also known as beige cells) in WAT. Recent studies have demonstrated that an increase in beige adipocytes in WAT enhances whole-body energy expenditure and is expected to reduce the risk of diet-induced obesity and metabolic diseases [25].
1.2.6. Mitochondrial Effects of the ABA/LANCL System
As mitochondria are the main energy producers in mammalian cells, it comes as no surprise that the ABA/LANCL system stimulates several mitochondrial functions, including mitochondrial biogenesis, oxidative metabolism, oxygen consumption, and proton gradient accumulation, in both adipose and muscle cells.
In murine and human preadipocytes, in vitro ABA does not induce triglyceride accumulation; instead, it increases the mitochondrial content, O2 consumption, and CO2 production sustained by LANCL2-dependent increased GLUT4 expression and glucose uptake and stimulates the transcription of “browning” genes, such as uncoupling protein-1 (UCP-1) [18]. In vivo, one-month-long ABA treatment at 1 µg/Kg BW/day significantly increased the mitochondrial DNA content in the WAT and in WAT-derived preadipocytes differentiated in vitro from treated mice compared with untreated controls. ABA also increased the expression of mitochondrial uncoupling proteins 1 and 3 (UCP-1/3) in brown adipose tissue from treated mice and stimulated BAT glucose uptake, an indirect measure of BAT thermogenesis [18].
In rodent L6 myocytes, LANCL1/2 overexpression and/or ABA treatment stimulated mitochondrial biogenesis, as witnessed by increased mitochondrial DNA and O2 consumption [19].
1.2.7. Non-Overlapping Roles of ABA and Insulin in Energy Metabolism
Based on the above results, a non-overlapping role for ABA and insulin in muscle and adipocyte metabolism can be envisaged. Insulin induces the phosphorylation of Akt, inhibiting AMPK and shifting the metabolic program from the starved to the fed state, activating glucose transport and metabolism, and storing excess energy abundance via glycogen, protein, and lipid synthesis. ABA instead induces the phosphorylation of AMPK, thereby activating the metabolic response to starvation and/or low glucose availability. This response includes the stimulation of glucose transport and oxidation for energy production, mitochondrial biogenesis, respiration, and uncoupling (Figure 2).
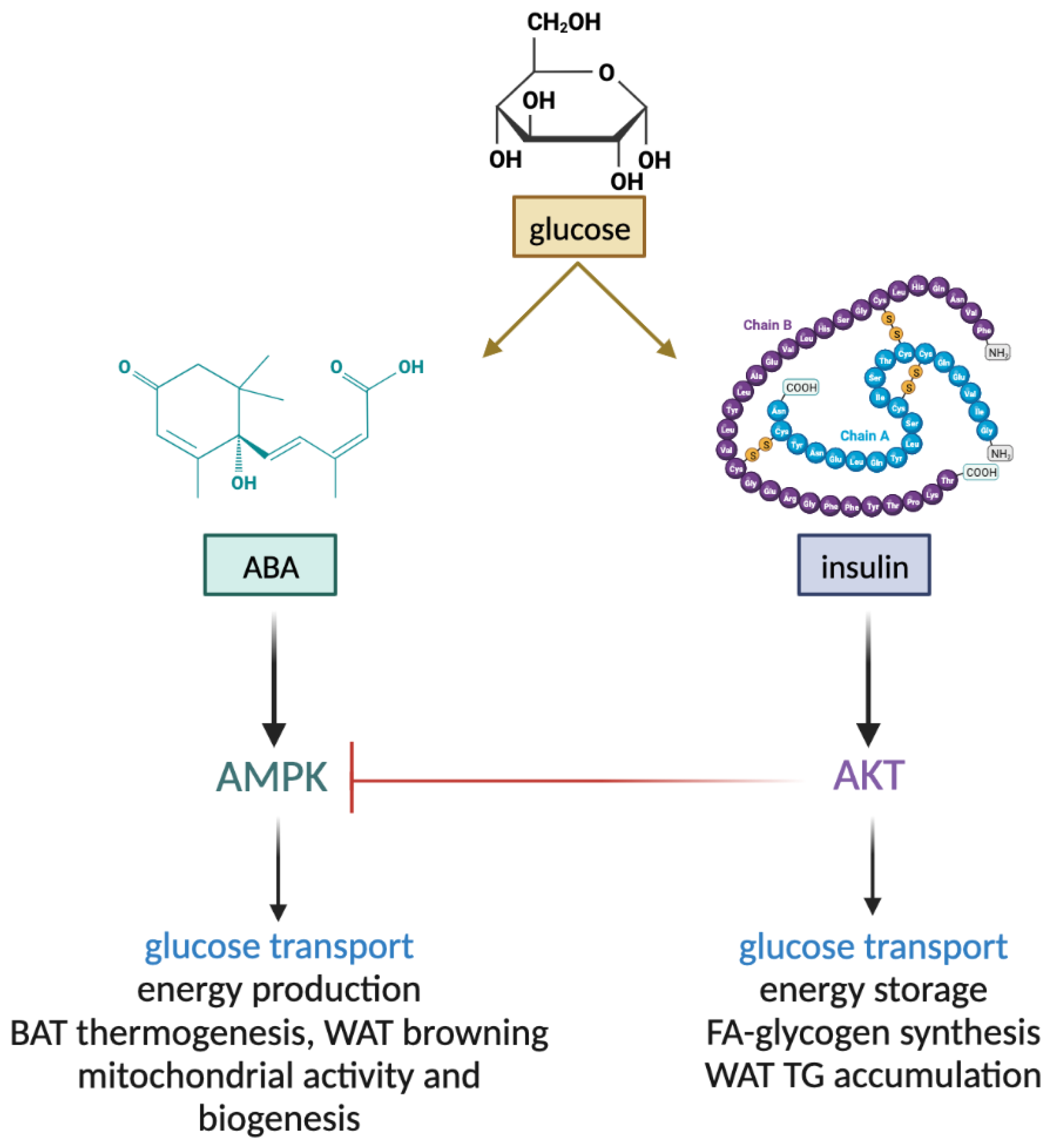
Figure 2. Non-overlapping functions of ABA and insulin in muscle and adipose tissue. ABA and insulin both stimulate glucose uptake by muscle and adipose tissue. Insulin, via Akt, stimulates the conversion of metabolic energy into storage forms, such as muscle glycogen and fatty acids and white adipocyte triglycerides. Activated Akt inhibits AMPK (blunted red line). Instead, ABA stimulates energy production via increased mitochondrial activity and biogenesis in muscle and adipose cells and thermogenesis. BAT, brown adipose tissue; WAT, white adipose tissue; FA, fatty acids; TG, triglycerides.
2. The ABA/LANCL System in Diabetes
2.1. Plasma ABA in Diabetic Subjects
In mammals, two peptide hormones are released by glucose/nutrient-sensing cells: pancreatic islet β-cells release insulin and intestinal endocrine cells release glucagon-like peptide 1 (GLP-1). Among other actions, GLP-1 contributes to stimulate insulin release and inhibits secretion by pancreatic α-cells of glucagon, the principal hormone activated by low blood glucose levels. ABAp also increases after an oral glucose load in healthy humans but not in subjects with type 2 diabetes (T2D) or with gestational diabetes (GDM) [26]. In GDM, resolution of the diabetic state that follows childbirth is accompanied by the restoration of normal ABAp, suggesting a critical role for ABAp in the maintenance of a normal glucose tolerance and a new possible ABA-centered pathogenetic mechanism that may underlie the diabetic condition. Indeed, the identification of a second hormone beside insulin capable of stimulating muscle glucose uptake would have significant consequences in diabetes mellitus, where insulin deficiency or insulin resistance reduce glucose tolerance. Together with insulin, ABAp is also undetectable or very low in type 1 diabetes (T1D) patients, suggesting that β-cells are the principal source of endogenous ABA in humans; thus, the demise of β-cells in T1D greatly reduces the availability of both hormones regulating glycemia, only one of which is currently replaced by therapy. As the metabolic actions of insulin and ABA are distinct, supplementation with one hormone cannot restore the functions of the other.
Indeed, low-dose oral ABA reduces glycemia and insulinemia in rats and in healthy humans undergoing a glucose load. The glycemia-lowering action of low-dose ABA in vivo reduces stimulation by hyperglycemia on β-cells and consequently insulin release. At variance with insulin, ABA does not induce hypoglycemia, even at a dose 100,000 times higher than the one efficacious in reducing glycemia (100 mg/Kg BW vs. 1 µg/Kg BW); thus, it has a very high therapeutic index; the absence of hypoglycemic risk due to excess dosage places ABA at significant variance with respect to insulin and to oral hypoglycemic drugs.
Intriguingly, the LANCL1 gene is located within the Insulin-dependent diabetes (Idd) 5.3 locus, which provides resistance to T1D in NOD mice [27]. LANCL1 is also among the candidate genes responsible for an observed complex phenotype of impaired neuronal function due to a microdeletion in the chromosomal region 2q34 [28].
ABA can be administered orally, it is readily absorbed because in the acidic gastric environment the protonated molecule is membrane permeant, and its plasma concentration remains high for several hours after intake [17], probably due to its binding to plasma proteins, which reduces renal clearance.
2.2. Clinical Studies on Borderline and Prediabetic Subjects
Indeed, clinical studies performed on healthy subjects with borderline values for metabolic syndrome [29] or prediabetes [24] indicate that low-dose ABA reduces glycemia, lipidemia, cardiovascular risk parameters, and low-grade inflammation after daily chronic administration.
In subjects with borderline values for metabolic syndrome (HbA1c and FPG values, TC, WC, and BMI) low-dose ABA supplementation (1 µg/Kg BW/day) for 75 days by means of a vegetal extract titrated in ABA reduced fasting glycemia and insulinemia, glycemia AUC after glucose load, HbA1c, TC, and body weight. As a consequence, the 10-year cardiovascular risk was significantly reduced [29]. In parallel with the human study, employing a vegetal extract as the source of ABA, mice fed a high-glucose diet were treated with the pure ABA molecule at the same daily dose as humans (1 µg/Kg BW/day) for four months, resulting in an improvement of glucose tolerance and a reduction of HbA1c, of blood lipids, and of body weight in the ABA-treated animals compared with untreated controls.
In prediabetic subjects (IFG or IGT) low-dose ABA supplementation improved glyco-metabolic and inflammation parameters [24]. ABA treatment did not significantly modify the anthropometric parameters, but a reduction of TC, LDL-C, and Tg was observed, indicating a similar downward trend as observed in borderline subjects. ABA supplementation also significantly reduced hs-CRP levels, demonstrating an improvement in the inflammatory status of prediabetic patients. Indeed, chronic low-grade inflammation lies at the heart of the pathogenetic mechanism underlying insulin resistance.
Based on the results of these studies, it is possible to conclude that the improvements in metabolic and bodily parameters observed with the food supplements were due to ABA in the vegetal extract of the compositions because similar results were observed in mice fed the pure molecule.
In another clinical set-up, a single dose of an ABA-containing vegetal extract was tested on the glycemia profile after intake of a standardized carbohydrate-rich breakfast. In each subject, breakfast with the food supplement significantly reduced the mean glycemia profile and the mean AUC of glycemia compared with the same breakfast without the supplement [29]. Ingestion of the ABA-rich extract increased ABAp 5- to 16-fold over fasting levels (5–15 nM), indicating that oral ABA is absorbed readily and contributes to the endogenous ABAp pool [17]. Reduction of post-prandial glycemia and insulinemia in normal subjects was also reported after intake of a different vegetal extract, titrated in ABA [30], indicating that the nature of the vegetal extract is irrelevant, as long as it contains a sufficient amount of ABA.
Together, these data outline an important role for the ABA/LANCL system in the physiology of glucose metabolism and energy metabolism in mammals. This background allows to hypothesize a beneficial role for ABA in conditions were glucose metabolism and/or mitochondrial energy production are deranged. The extent to which insulin and ABA synergize to control glycemia is an open field of investigation. In both T1D and T2D, a severe reduction of endogenous plasma ABA or of the plasma ABA response to hyperglycemia occurs [26]. ABA appears to be mainly produced by β-cells, as plasma ABA is very low or undetectable in T1D subjects. Another possible explanation for this observation is that insulin promotes the release of ABA (also) from other cell types; in any case, a severe reduction or the complete destruction of β-cells could affect the release of both hormones uniquely endowed with the ability to stimulate muscle glucose uptake.
2.3. Oral ABA Ameliorates Glycemia in Insulin-Deficient Mice
The genetic ablation of Ca2+-permeable non-selective cation channel TRPM2 results in defective insulin secretion in CD1 mice; consequently, TRPM2 KO mice fed a high-glucose diet develop hyperglycemia due to insufficient insulin release [31]. In hypoinsulinemic TRPM2 KO mice, treatment with ABA, both at a single dose together with a glucose load, or chronically, in high-glucose-fed mice, reduced the glycemia profile and increased muscle glycogen storage without increasing plasma insulin levels [20].
Another murine model of insulin deficiency is the STZ-induced β-cell loss that mimics the demise of the β cell reservoir occurring due to autoimmune aggression in T1D. This experimental model showed a gradual loss of β cells over several weeks, or instead, a rapid induction of almost total β cell loss and consequent hyperglycemia over a short period of days depending on the dose and timing of STZ administration, i.e., multiple low-dose STZ (MLD) or single high-dose STZ (SHD). Recent studies have tested that treatment with ABA could improve glycemic control in these murine models of T1D, either alone, during the progressive β cell loss induced by MLD-STZ, or in addition to insulin, under conditions of complete endogenous insulin deficiency (SHD-STZ). The MLD and SHD-STZ protocols mimic the relative insulin deficiency observed in T2D and the absolute insulin deficiency of T1D, respectively.
In the MLD protocol of T1D induction, chronic ABA treatment improved the glycemic profile in treated mice compared with untreated controls during a 28-day period without a significant difference between plasma insulin levels after a final OGTT and with similar residual amounts of pancreatic insulin mRNA at the end-point. Thus, the improvement of glycemic control in the ABA-treated animals was not attributed to higher endogenous insulin levels but rather to the glycemia-lowering action of ABA via an increased skeletal muscle glucose uptake. Furthermore, ABA-treated mice had increased expression of the insulin receptor in skeletal muscle, suggesting an improved action of residual endogenous insulin [32].
In the SHD protocol, ABA alone could not substitute for insulin under conditions of total insulin deficiency when glycemia increased to higher than 500 mg/dL, but it improved the effect of exogenous insulin, when the dose of the peptide hormone was insufficient to restore euglycemia [32].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24021199
References
- Bruzzone, S.; Moreschi, I.; Usai, C.; Guida, L.; Damonte, G.; Salis, A.; Scarfì, S.; Millo, E.; De Flora, A.; Zocchi, E. Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc. Natl. Acad. Sci. USA 2007, 104, 5759–5764.
- Desikan, R.; Cheung, M.K.; Bright, J.; Henson, D.; Hancock, J.T.; Neill, S.J. ABA, hydrogen peroxide and nitric oxide signalling in stomatal guard cells. J. Exp. Bot. 2004, 55, 205–212.
- Bruzzone, S.; Basile, G.; Mannino, E.; Sturla, L.; Magnone, M.; Grozio, A.; Salis, A.; Fresia, C.; Vigliarolo, T.; Guida, L.; et al. Autocrine Abscisic Acid Mediates the UV-B-Induced Inflammatory Response in Human Granulocytes and Keratinocytes. J. Cell Physiol. 2012, 227, 2502–2510.
- Tossi, V.; Cassia, R.; Bruzzone, S.; Zocchi, E.; Lamattina, L. ABA Says NO to UV-B: A Universal Response? Trends Plant Sci. 2012, 17, 510–517.
- Sun, Y.; Pri-Tal, O.; Michaeli, D.; Mosquna, A. Evolution of Abscisic Acid Signaling Module and Its Perception. Front. Plant Sci. 2020, 11, 934.
- Bauer, H.; Mayer, H.; Marchler-Bauer, A.; Salzer, U.; Prohaska, R. Characterization of P40/GPR69A as a Peripheral Membrane Protein Related to the Lantibiotic Synthetase Component C. Biochem. Biophys. Res. Commun. 2000, 275, 69–74.
- Landlinger, C.; Salzer, U.; Prohaska, R. Myristoylation of Human LanC-like Protein 2 (LANCL2) Is Essential for the Interaction with the Plasma Membrane and the Increase in Cellular Sensitivity to Adriamycin. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1759–1767.
- He, C.; Zeng, M.; Dutta, D.; Koh, T.H.; Chen, J.; van der Donk, W.A. LanCL Proteins Are Not Involved in Lanthionine Synthesis in Mammals. Sci. Rep. 2017, 7, 40980.
- Lai, K.-Y.; Galan, S.R.G.; Zeng, Y.; Zhou, T.H.; He, C.; Raj, R.; Riedl, J.; Liu, S.; Chooi, K.P.; Garg, N.; et al. LanCLs Add Glutathione to Dehydroamino Acids Generated at Phosphorylated Sites in the Proteome. Cell 2021, 184, 2680–2695.e26.
- Liu, X.; Yue, Y.; Li, B.; Nie, Y.; Li, W.; Wu, W.H.; Ma, L. A G Protein-Coupled Receptor Is a Plasma Membrane Receptor for the Plant Hormone Abscisic Acid. Science 2007, 315, 1712–1716.
- Pandey, S.; Nelson, D.C.; Assmann, S.M. Two Novel GPCR-Type G Proteins Are Abscisic Acid Receptors in Arabidopsis. Cell 2009, 136, 136–148.
- Kharenko, O.A.; Choudhary, P.; Loewen, M.C. Abscisic Acid Binds to Recombinant Arabidopsis Thaliana G-Protein Coupled Receptor-Type G-Protein 1 in Sacaromycese Cerevisiae and in Vitro. Plant Physiol. Biochem. 2013, 68, 32–36.
- Cichero, E.; Fresia, C.; Guida, L.; Booz, V.; Millo, E.; Scotti, C.; Iamele, L.; de Jonge, H.; Galante, D.; De Flora, A.; et al. Identification of a High Affinity Binding Site for Abscisic Acid on Human Lanthionine Synthetase Component C-like Protein 2. Int. J. Biochem. Cell Biol. 2018, 97, 52–61.
- Sturla, L.; Fresia, C.; Guida, L.; Bruzzone, S.; Scarfì, S.; Usai, C.; Fruscione, F.; Magnone, M.; Millo, E.; Basile, G.; et al. LANCL2 Is Necessary for Abscisic Acid Binding and Signaling in Human Granulocytes and in Rat Insulinoma Cells. J. Biol. Chem. 2009, 284, 28045–28057.
- Fresia, C.; Vigliarolo, T.; Guida, L.; Booz, V.; Bruzzone, S.; Sturla, L.; Di Bona, M.; Pesce, M.; Usai, C.; De Flora, A.; et al. G-Protein Coupling and Nuclear Translocation of the Human Abscisic Acid Receptor LANCL2. Sci. Rep. 2016, 6, 26658.
- Vigliarolo, T.; Zocchi, E.; Fresia, C.; Booz, V.; Guida, L. Abscisic Acid Influx into Human Nucleated Cells Occurs through the Anion Exchanger AE2. Int. J. Biochem. Cell Biol. 2016, 75, 99–103.
- Magnone, M.; Ameri, P.; Salis, A.; Andraghetti, G.; Emionite, L.; Murialdo, G.; De Flora, A.; Zocchi, E. Microgram Amounts of Abscisic Acid in Fruit Extracts Improve Glucose Tolerance and Reduce Insulinemia in Rats and in Humans. FASEB J. 2015, 29, 4783–4793.
- Sturla, L.; Mannino, E.; Scarfì, S.; Bruzzone, S.; Magnone, M.; Sociali, G.; Booz, V.; Guida, L.; Vigliarolo, T.; Fresia, C.; et al. Abscisic Acid Enhances Glucose Disposal and Induces Brown Fat Activity in Adipocytes in Vitro and in Vivo. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 131–144.
- Spinelli, S.; Begani, G.; Guida, L.; Magnone, M.; Galante, D.; D’Arrigo, C.; Scotti, C.; Iamele, L.; De Jonge, H.; Zocchi, E.; et al. LANCL1 Binds Abscisic Acid and Stimulates Glucose Transport and Mitochondrial Respiration in Muscle Cells via the AMPK/PGC-1α/Sirt1 Pathway. Mol. Metab. 2021, 53, 101263.
- Magnone, M.; Emionite, L.; Guida, L.; Vigliarolo, T.; Sturla, L.; Spinelli, S.; Buschiazzo, A.; Marini, C.; Sambuceti, G.; De Flora, A.; et al. Insulin-Independent Stimulation of Skeletal Muscle Glucose Uptake by Low-Dose Abscisic Acid via AMPK Activation. Sci. Rep. 2020, 10, 1454.
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin Antagonizes Ischemia-Induced Thr172 Phosphorylation of AMP-Activated Protein Kinase α-Subunits in Heart via Hierarchical Phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340.
- Valentine, R.J.; Coughlan, K.A.; Ruderman, N.B.; Saha, A.K. Insulin Inhibits AMPK Activity and Phosphorylates AMPK Ser485/491 through Akt in Hepatocytes, Myotubes and Incubated Rat Skeletal Muscle. Arch. Biochem. Biophys. 2014, 562, 62–69.
- Guri, A.J.; Hontecillas, R.; Si, H.; Liu, D.; Bassaganya-Riera, J. Dietary Abscisic Acid Ameliorates Glucose Tolerance and Obesity-Related Inflammation in Db/Db Mice Fed High-Fat Diets. Clin. Nutr. 2007, 26, 107–116.
- Derosa, G.; Maffioli, P.; D’Angelo, A.; Preti, P.S.; Tenore, G.; Novellino, E. Abscisic Acid Treatment in Patients with Prediabetes. Nutrients 2020, 12, 2931.
- Pan, R.; Zhu, X.; Maretich, P.; Chen, Y. Metabolic Improvement via Enhancing Thermogenic Fat-Mediated Non-Shivering Thermogenesis: From Rodents to Humans. Front. Endocrinol. 2020, 11, 633.
- Ameri, P.; Bruzzone, S.; Mannino, E.; Sociali, G.; Andraghetti, G.; Salis, A.; Ponta, M.L.; Briatore, L.; Adami, G.F.; Ferraiolo, A.; et al. Impaired Increase of Plasma Abscisic Acid in Response to Oral Glucose Load in Type 2 Diabetes and in Gestational Diabetes. PLoS ONE 2015, 10, e0115992.
- Hunter, K.; Rainbow, D.; Plagnol, V.; Todd, J.A.; Peterson, L.B.; Wicker, L.S. Interactions between Idd5.1/Ctla4 and Other Type 1 Diabetes Genes. J. Immunol. 2007, 179, 8341–8349.
- Westphal, D.S.; Andres, S.; Makowski, C.; Meitinger, T.; Hoefele, J. MAP2—A Candidate Gene for Epilepsy, Developmental Delay and Behavioral Abnormalities in a Patient with Microdeletion 2q34. Front. Genet. 2018, 9, 99.
- Magnone, M.; Leoncini, G.; Vigliarolo, T.; Emionite, L.; Sturla, L.; Zocchi, E.; Murialdo, G. Chronic Intake of Micrograms of Abscisic Acid Improves Glycemia and Lipidemia in a Human Study and in High-Glucose Fed Mice. Nutrients 2018, 10, 1495.
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Goodpaster, B.; Bassaganya-Riera, J. Abscisic Acid Enriched Fig Extract Promotes Insulin Sensitivity by Decreasing Systemic Inflammation and Activating LANCL2 in Skeletal Muscle. Sci. Rep. 2020, 10, 10463.
- Uchida, K.; Dezaki, K.; Damdindorj, B.; Inada, H.; Shiuchi, T.; Mori, Y.; Yada, T.; Minokoshi, Y.; Tominaga, M. Lack of TRPM2 Impaired Insulin Secretion and Glucose Metabolisms in Mice. Diabetes 2011, 60, 119–126.
- Magnone, M.; Spinelli, S.; Begani, G.; Guida, L.; Sturla, L.; Emionite, L.; Zocchi, E. Abscisic Acid Improves Insulin Action on Glycemia in Insulin-Deficient Mouse Models of Type 1 Diabetes. Metabolites 2022, 12, 523.
This entry is offline, you can click here to edit this entry!