Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Azomethine ylides are nitrogen-based three-atom components commonly used in [3+2]-cycloaddition reactions with various unsaturated 2π-electron components. These reactions are highly regio- and stereoselective and have attracted the attention of organic chemists with respect to the construction of diverse heterocycles potentially bearing four new contiguous stereogenic centers.
- cycloaddition
- azomethine ylide
- pyrrolidine
1. Introduction
The three-atom component (TAC) is an organic species that is represented by zwitterionic octet structures and undergoes [3+2]-cycloadditions with an unsaturated 2π-electron component in a one-step reaction, often in an asynchronous and symmetry-conducive fashion, via a thermal six-electron Hückel aromatic transition state. The formal charges are lost in the [3+2→5] cycloaddition [1]. Recently, studies based on molecular electron density theory (MEDT) have suggested that the compounds involved in these reactions do not have a polar nature but a diradical, pseudoradical, or carbenoid nature. Therefore, the use of the term “1,3-dipole” is unjustified and should be replaced with “three-atom component”. It was also recommend that the designation of “dipolarophile” should be replaced with “unsaturated 2π-electron component”, and “1,3-dipolar cycloaddition” with “[3+2]-cycloaddition” [2].
While there is a mechanistic spectrum of this reaction from a synchronous one-step process to a stepwise overall transformation (including radical pathways), to avoid mechanistic digressions that may not have chemical or stereochemical consequences, the azomethine ylide reaction will be refered to as a pericyclic cycloaddition. [3+2]-Cycloadditions of azomethine ylide with homomultiple and heteromultiple unsaturated 2π-electron components have been extensively used to produce a wide range of heterocycles [3]. There are several methods for the formation of azomethine ylides, including the thermolysis or photolysis of readily prepared aziridines, the dehydrohalogenation of immonium salts, and proton abstraction from imine derivatives of α-amino acids [3]. They are often generated in situ because of their high reactivity and/or transient existence; however, in some cases, stabilized ylides have been isolated and used further [4][5][6].
The synthesis of five-membered heterocyclic systems through azomethine ylides is one of the most adopted, efficient, and powerful approaches. Since the first report of successful the enantioselective [3+2]-cycloaddition of an azomethine ylide in 1991 [7], there has been tremendous progress in the chemistry regarding azomethine ylides. Azomethine ylides are extensively used in the synthesis of various heterocyclic systems such as pyrrolidines, pyrrolizidines, indolizidines, piperidines, oxazolidines, spiroindoles, spiropyrrolidines, and spiropiperidines, but they are also used for the total synthesis of complex natural products as well as bioactive compounds [8][9][10][11][12][13][14][15]. In recent years, the [3+2]-cycloaddition reaction has been extensively studied for the synthesis of heterocycles using different synthetic strategies [16][17]. In addition, the reaction is also investigated to understand the related reactivity, reaction conditions, intermediates, etc. [18][19].
2. Intermolecular Cycloaddition Reaction of Azomethine Ylides to Acyclic Unsaturated 2π-Electron Components (Alkenes)
Unstabilized azomethine ylide 2 derived from benzyl(methoxymethyl)(trimethylsilylmethyl)amine 1 undergoes a [3+2]-cycloaddition reaction with electron-deficient alkenes 3 under continuous flow conditions in the presence of catalytic trifluoroacetic acid, thereby affording the corresponding pyrrolidines 4 (Scheme 1) [20].
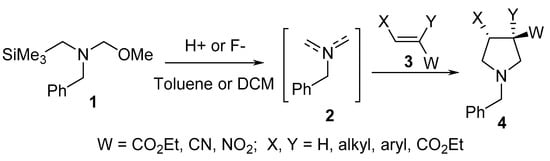
Scheme 1. Synthesis of pyrrolidines 4.
Azomethine ylides generated via the deprotonation of α-imino-esters 5 undergo a [3+2]-cycloaddition reaction with unsaturated 2π-electron components 6 in the presence of the eco-friendly supported solid-base catalyst KF/Al2O3 to yield the corresponding pyrrolidines 7 with high regio- and diastereoselectivity (Scheme 2) [21].
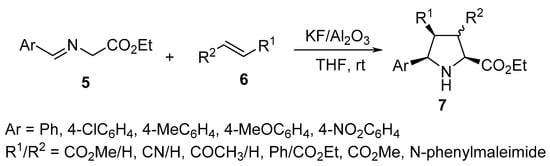
Scheme 2. Synthesis of pyrrolidines 7.
Belfaitah et al. reported the cycloaddition reaction of azomethine ylides 9 with alkenyl boronates 8 to obtain the 3-boronic-ester-substituted pyrrolidines 10 (Scheme 3) [22].
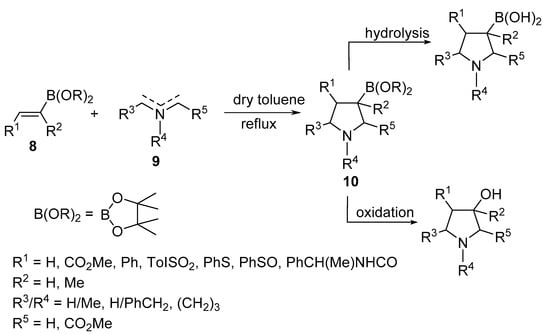
Scheme 3. Synthesis of 3-boronate pyrrolidines 10.
Pyrrolo[2,1-a]isoquinolines 15 were obtained through a sequential one-pot, two-step tandem reaction of isoquinoline 11, α-halogenated methylenes 12, aromatic aldehydes 13, and cyanoacetoamide 14 in the presence of triethylamine as a basic catalyst and 2,4-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as an oxidizing agent. The transformation was assumed to take place through [3+2]-cycloaddition of N-substituted carbonylmethyleneisoquinolinium bromide (formed via the reaction of isoquinoline 11 and 12) with arylidene cyanoacetamide (formed via the condensation of cyanoacetamide 14 with aromatic aldehyde 13) [23]. In the case of the ethyl bromoacetate 16 derivative, the formation of pyrrolo[2,1-a]isoquinolines 17 was observed probably due to DDQ oxidation (Scheme 4) [23].
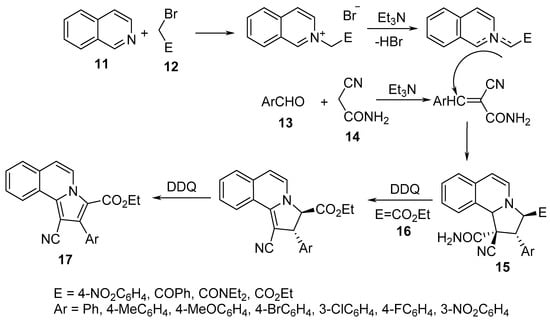
Scheme 4. Synthesis of pyrrolo[2,1-a]isoquinolines 15/17.
Spiro[indoline-3,2′-pyrrolidines] 21 were prepared by the [3+2]-cycloaddition reaction of benzoimidazol-2-yl-3-phenylacrylonitriles 18 with azomethine ylides, which was generated in situ from the condensation of isatin 19 and sarcosine 20 in refluxing ethanol. Similarly, spiro[indoline-3,5′-pyrrolo[1,2-c]thiazoles] 23 were formed by using thioproline 22 as a secondary amino acid (Scheme 5) [24].
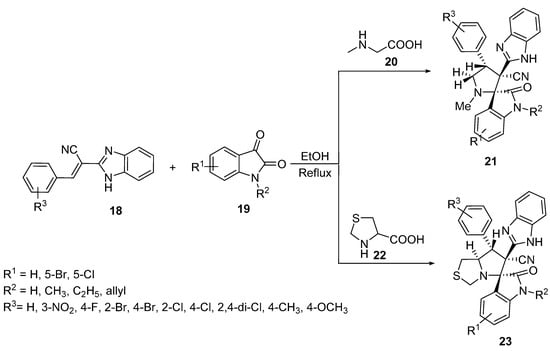
Scheme 5. Synthesis of spiro[indoline-3,2′-pyrrolidines] 21 and spiro[indoline-3,5′-pyrrolo[1,2-c]thiazoles] 23.
The chemistry was extended further to obtain spiro[acenaphthylene-1,2′-pyrrolidines] 26 and spiro[acenaphthylene-1,2′-pyrrolizidines] 28 possessing a cyano group from the azomethine ylides (generated from acenaphthenequinone 25) with α-amino acids (sarcosine 20 and proline 27) and Knoevenagel adducts 24 (Scheme 6) [25].
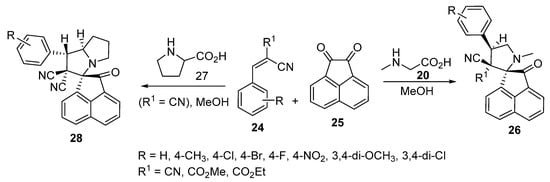
Scheme 6. Synthesis of spiro[acenaphthylene-1,2′-pyrrolidines] 26 and spiro[acenaphthylene-1,2′-pyrrolizidines] 28.
3. Nitroalkenes
Nitroalkenes are reactive, unsaturated 2π-electron components that are intensively used in cycloaddition reactions by various researchers [26]. 3-Nitro-4-(trichloromethyl)pyrrolidine 30 was obtained through the cycloaddition of trans-3,3,3-trichloro-1-nitroprop-1-ene 29 with azomethine ylide (obtained from the condensation of paraformaldehyde and sarcosine in refluxing benzene). Quantum chemical calculations (DFT, M062X/6-311G(d)) explained the reaction pathway [27]. Analogously, 3-nitro-4-arylpyrrolidine-3-carbonitriles 32 were obtained through the cycloaddition of the azomethine ylide with (2E)-3-phenyl-2-nitroprop-2-enenitriles 31 [28] (Scheme 7).
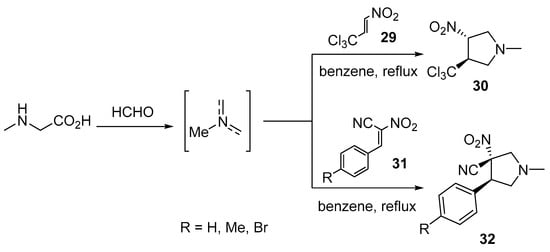
Scheme 7. Synthesis of 3-nitro-4-(trichloromethyl)pyrrolidine 30 and 3-nitro-4-arylpyrrolidine-3-carbonitriles 32.
Trans-3-nitropyrrolidine 34 was prepared by reacting trans-1-nitro-2-phenylethylene 33 with N-(methoxymethyl)-N-[(trimethylsilyl)methyl]benzylamine 1, which is an azomethine ylide equivalent, in the presence of trifluoroacetic acid in dichloromethane. Some of the synthesized 34 revealed promising inhibitory properties as Na+ channel blockers, which are useful in the treatment of ischemic stroke (Scheme 8) [29].
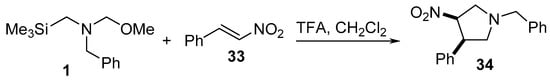
Scheme 8. Synthesis of trans-3-nitropyrrolidine 34.
Another set of spiro compounds, spiro[pyrrolidine-2,3′-oxindoles] 37, were regioselectively synthesized by a multicomponent reaction of azomethine ylides, generated in situ from 3-aminoindoline-2-ones hydrochloride 35, with aldehydes 13 and (E)-nitroalkenes 36 (Scheme 9) [30].
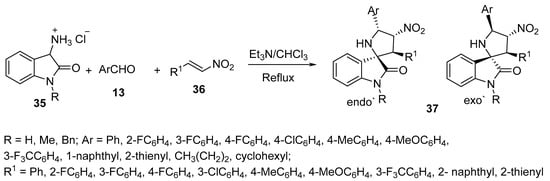
Scheme 9. Synthesis of spiro[pyrrolidin-2,3′-oxindoles] 37.
It was assumed that, based on the secondary orbital interaction (SOI) of the electron-poor nitroalkenes 36 with the azomethine ylide, Path A was exclusively followed, as the endo-transition state in the reaction sequence was more energetically favorable (Scheme 10) [30].
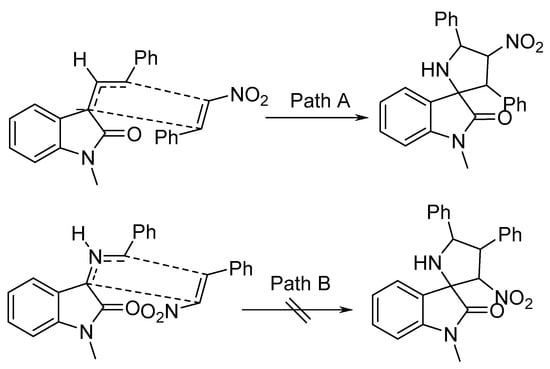
Scheme 10. Proposed mechanism for the cycloaddition of azomethine ylide (via endo′-transition state).
Spirooxindolo-nitropyrrolizines 38 (major product) and 39 (minor product) were obtained from the cycloaddition reaction of azomethine ylides, generated in situ from isatin 19, with proline 27 and (E)-ß-nitrostyrene 32 (Scheme 11) [31]. A significant inversion in the regioselectivity was observed when the polar [3+2]-cycloaddition of the azomethine ylides was attempted with trans-β-nitrostyrene instead of (E)-1-phenyl-2-nitropropene.
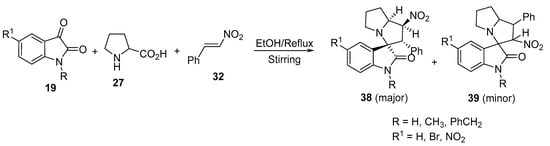
Scheme 11. Synthesis of spirooxindolo-nitropyrrolizines 38 and 39.
It was assumed that the reaction proceeds through S-shaped ylide with a cycloaddition via the endo-transition state (pathway B), yielding cycloadducts 38, and not the exo-transition state (pathway A). Computational studies (Gaussian 03) of the transition states (Density Functional Theory (DFT), B3LYP, and 6-31G(d,p) basis set) confirmed these assumptions (Scheme 12) [31].
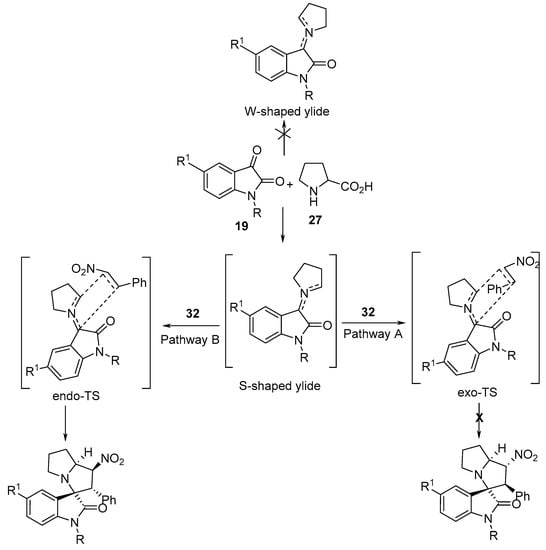
Scheme 12. Proposed mechanism for the cycloaddition of the azomethine ylides with nitrostyrene.
A series of spiro[indoline-3,3′-pyrrolizin]-2-ones 40 with potential anti-amyloidogenic properties useful against Alzheimer’s disease were obtained by the microwave-assisted cycloaddition of nitroalkenes 36 and azomethine ylides (generated from isatin 19 and L-proline 27) [32]. Analogously, spirooxindole-pyrrolidines 42 were obtained by the reaction of tyrosine 41 in an ionic liquid [bmim]Br at 100 °C. Promising antiproliferation properties were observed for some of the synthesized compounds (42) against human A549 (adenocarcinoma basal epithelial) and Jurkat (T-cell lymphoma) cell lines (MTT assay) using Camptothecin as a positive control; the compounds exhibited a safe response against the non-cancer cell lines MCF-10 (normal breast) and PCS-130-010 (lung smooth muscle). Caspase-dependent apoptosis (especially caspase-3) was mentioned as the mode of action for the observed antiproliferative activity (Scheme 13) [33].
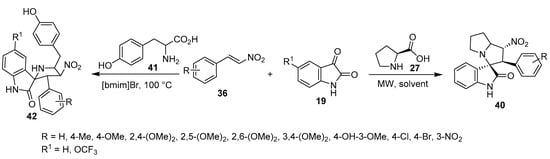
Scheme 13. Synthesis of spiro-indolines 40, 42.
Ionic liquid chemistry was utilized to prepare 4′-nitrospiro[indeno[1,2-b]quinoxaline-11,2′-pyrrolidines] 47 by the cycloaddition reaction of nitroalkenes 36 with azomethine ylide (generated from indenoquinoxalinone 45 and L-phenylalanine 46) in an ionic liquid [bmim]Br. Some of the synthesized agents revealed antimycobacterial properties (Mycobacterium tuberculosis H37Rv) with an efficacy comparable to that of ethambutol (reference standard) [34]. Similarly, spiro compounds 49 were obtained by using L-histidine 48 instead of L-phenylalanine 46 in this reaction. Some of the synthesized compounds revealed cholinesterase (acetylcholinesterase and butyrylcholinesterase)-inhibitory properties with considerable efficiencies relative to Galantamine (Scheme 14) [35].
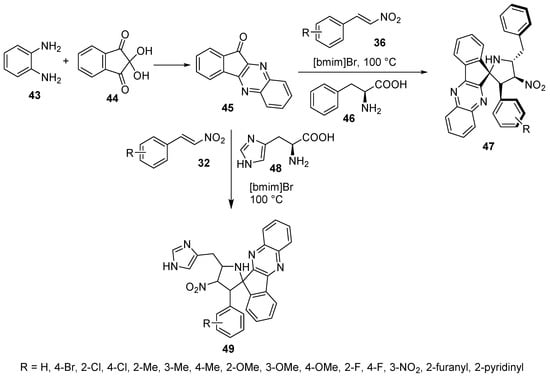
Scheme 14. Synthesis of 4′-nitrospiro[indeno[1,2-b]quinoxaline-11,2′-pyrrolidines] 47, 49.
Pyrrolidinyl ß-lactams 52 were prepared as single diastereomers by the reaction of azomethine ylides 51, generated from β-lactam imines of α-amino ester 50, with nitrostyrenes 36 in the presence of silver acetate and triethylamine (Scheme 15). This reaction is an example of [3+2]-cycloaddition reaction via N-metallo azomethine ylide [36].
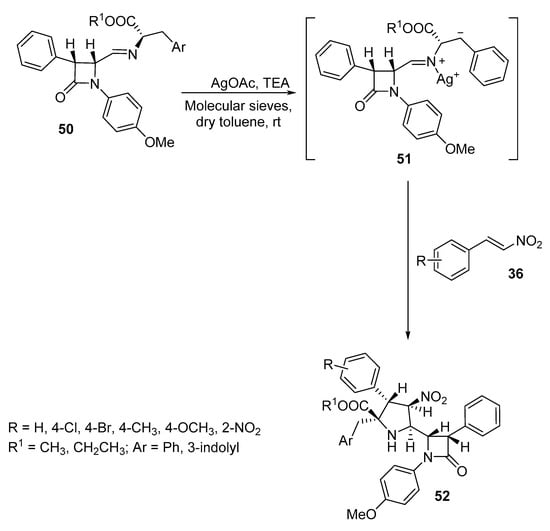
Scheme 15. Synthesis of pyrrolidinyl β-lactams 52.
3,4-Dihydropyrrolo[2,1-a]isoquinolines 54 were obtained by the [3+2]-cycloaddition reaction of nitroalkenes 36 with an azomethine ylide that was efficiently generated via the dirhodium(II)caprolactamate [Rh2(cap)4] catalyzed oxidation of tetrahydroisoquinoline 53 (Scheme 16). Doyle’s oxidative protocol was used to generate azomethine ylides, which were further trapped in situ via [3+2]-cycloaddition [37].
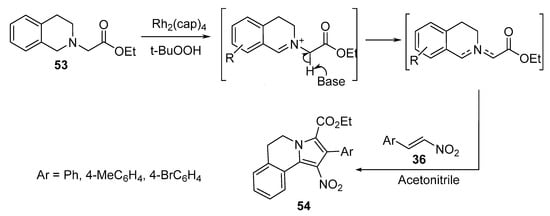
Scheme 16. Synthesis of pyrrolo[2,1-a]isoquinolines 54.
4. α,β-Unsaturated Polarophiles
Spiro[3H-indole-3,3′-[3H]pyrrolizin]-2-ones 56 were synthesized by the cycloaddition reaction of (E)-3-aryl-1-(thiophen-2-yl)-prop-2-en-1-ones 55 with azomethine ylide generated in situ from the condensation of isatin 19 with L-proline 27 (Scheme 17). Some of the synthesized spiroindoles 56 showed potential antibacterial activity against Staphylococcus aureus and Salmonella typhi (relative to Streptomycin) and antifungal activity against Candida albicans (relative to Amphotericin B) [38].
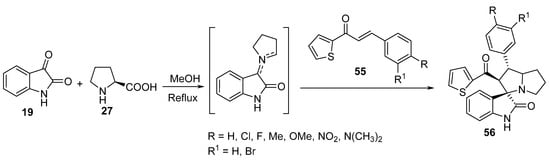
Scheme 17. Synthesis of spiro[3H-indole-3,3′-[3H]pyrrolizin]-2-ones 56.
Spiro[pyrrolidine-2,3′-indolin]-2′-ones 59 were synthesized by the multi-component cycloaddition reaction of chalcones 58 and an azomethine ylide formed from the condensation of isatin 19 and benzylaminemine 57. Few of the synthesized spiro-analogs 59 revealed potent inhibitory advanced glycation end (AGE) product formation in a bovine serum albumin (BSA)-glucose assay that was higher than that of aminoguanidine (standard reference). The occurrence of AGE is related to hyperglycemia observed as a complication of diabetes (Scheme 18) [39].
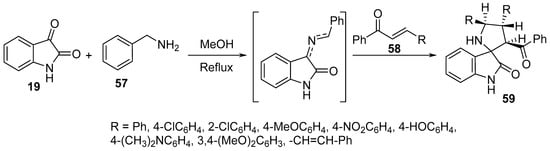
Scheme 18. Synthesis of spiro[pyrrolidine-2,3′-indolin]-2′-ones 59.
Taghizadeh et al. reported an efficient and greener multicomponent protocol for the synthesis of regio-, diastereo-, and enantioselective spiro-oxindolopyrrolizidines 61 from optically active cinnamoyl oxazolidinone 60 and azomethine ylides that were formed from the condensation reaction of isatin 19 and S-proline 27 (Scheme 19) [40].
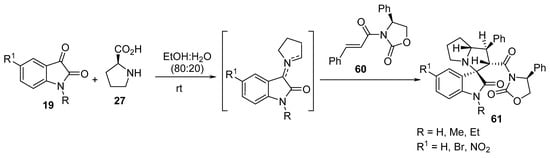
Scheme 19. Synthesis of the spiro-oxindolopyrrolizidines 61.
Spiro[indoline-3,2′-pyrrolidines] 63 were prepared by the reaction of compound 62 containing an α,β-unsaturated ketone function with azomethine ylides obtained from isatin 19 and sarcosine 20, while spiro[indoline-3,5′-pyrrolo[1,2-c]thiazoles] 64 was obtained from a similar reaction that involved thioproline 22 instead of sarcosine 20 (Scheme 20). Some of the synthesized spiro-compounds, 63 and 64, revealed anticancer properties against the A549 lung cancer cell line (MTT assay) [41][42] and spiro-compound 63 also showed antimicrobial activity against Gram-positive (Micrococcus luteus, Enterobacter aerogenes, Staphylococcus aureus and Staphylococcus aureus “MRSA-methicillin resistant”) and Gram-negative (Salmonella typhimurium, Klebsiella pneumoniae, Proteus vulgaris, and Shigella flexneri) bacterial strains and fungi (Malassesia pachydermatis, Candida albicans) relative to Streptomycin and Ketoconazole (used as antibacterial and antifungal standard references, respectively) [42].
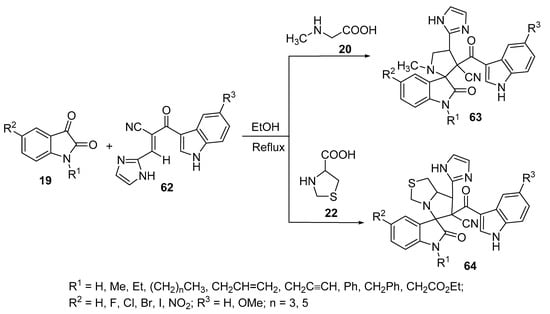
Scheme 20. Synthesis of spiro[indoline-3,2′-pyrrolidines] 63 and spiro[indoline-3,5′-pyrrolo[1,2-c]thiazoles] 64.
Spiropyrrolidine-oxindoles 66 were prepared in appreciable yields by the cycloaddition reaction of the unsaturated 2π-electron component (E)-2-(1H-indole-3-carbonyl)-3-phenylacrylonitrile 65 and azomethine ylides obtained from the condensation of isatin 19 and sarcosine 20 (Scheme 21) [43].
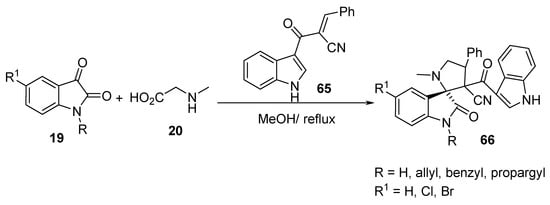
Scheme 21. Synthesis of spiropyrrolidine-oxindoles 66.
Similarly, spiropyrrolidine–oxindoles 68–70 were obtained from the reaction of enone 67 with azomethine ylides derived from isatin 19 and α-amino acids (sarcosine 20, proline 27 or thioproline 22). Among all the synthesized compounds, some showed antimicrobial properties against Gram-positive and Gram-negative bacterial as well as fungal strains using Streptomycin and Ketconazole as standard references (Scheme 22) [44].
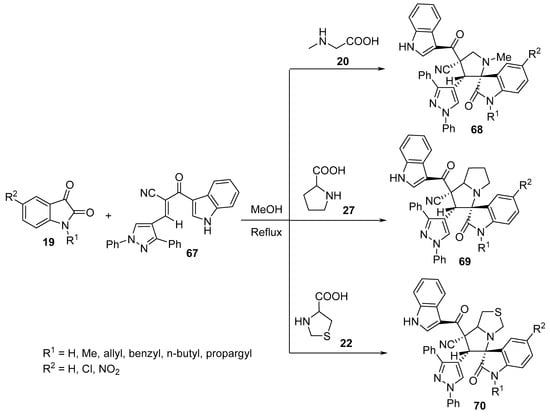
Scheme 22. Synthesis of spiropyrrolidine-oxindoles 68–70.
The unsaturated 2π-electron component, 2-[hydroxyl(4-oxo-4H-chromen-3-yl)methyl]acrylonitrile 71, was synthesized by the Baylis–Hillman reaction of chromene-3-aldehyde, treated with the azomethine ylides (from isatin 19 and sarcosine 20), which afforded the corresponding regioselective spiro[pyrrolidine-oxindoles] 72 and 73 as major and minor products, respectively (Scheme 23) [45].
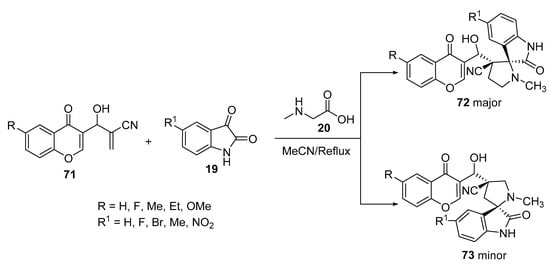
Scheme 23. Synthesis of spiro[pyrrolidine-oxindoles] 73, 74.
A convenient method for the selective construction of spiroindane-1,3-diones 77 relies upon the generation of unstabilized azomethine ylides from the initial condensation between ninhydrin 44 and 1,2,3,4-tetrahydroisoquinoline 74. Subsequent azomethine ylide cycloaddition onto the conjugated double bond of chalcone 76 was exploited, giving target cycloadducts with good yields (77–94%) and diastereoselectivity (Scheme 24) [46].
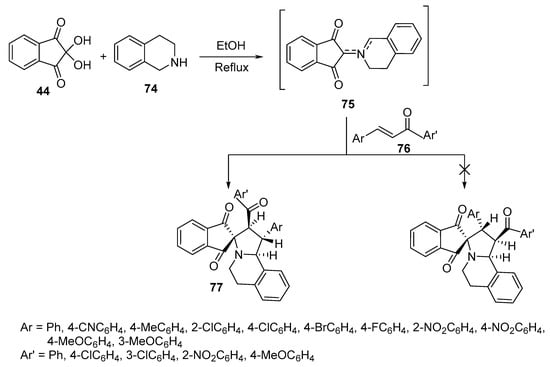
Scheme 24. Synthesis of spiroindane-1,3-diones 77.
The reaction of azomethine ylide generated from 5-choloroisatin 19 and L-proline 27 as well as 1-acryloyl-4-piperidinones 78 yielded the corresponding spirooxindole-pyrrolizines 79 (yield 62–84%). Some of the synthesized cycloadducts 79 displayed cholinesterase-inhibitory properties (acetylcholinesterase and butyrylcholinestrase) with potency relative to Galantamine [47]. When the reaction was conducted in a 1:2:2 molar ratio of 1-acryloyl-4-piperidinones 78, isatin 19, and L-proline 27, respectively, the bisspiropyrrolizines 80 were formed instead (yield 53–74%). It was found that most of the mono-spiropyrrolizines 79 (obtained using a 1:1:1 molar ratio of the reactants in yields of 73–84%) revealed higher cholinesterase enzyme (acetylcholinesterase and butyrylcholinestrase)-inhibitory activity than the bisspiropyrrolizine derivatives 80 (Scheme 25) [48].
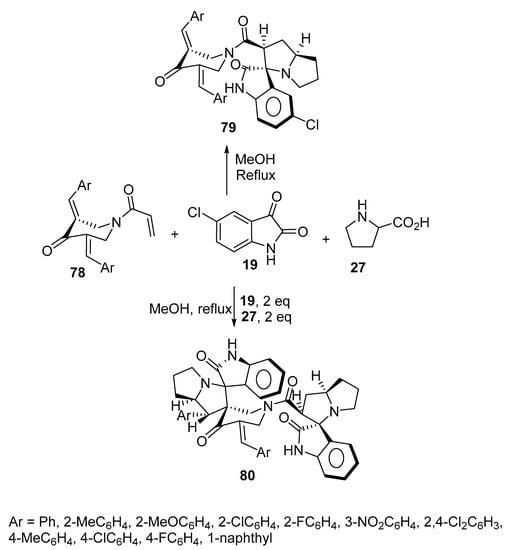
Scheme 25. Synthesis of mono-spiropyrrolizines 79 and bisspiropyrrolizines 80.
The reaction of 3-(3-phenylazetidin-2-yl) acrylates 81 with azomethine ylide formed by the condensation of ninhydrin 44 and amino acids (sarcosine 20/L-proline 27) afforded the corresponding spiroindanopyrrolidines 82 and spiroindanopyrrolizines 83 (Scheme 26). The synthesized cycloadducts 82 and 83 showed antibacterial properties against Proteus mirabilis, Proteus vulgaris, Salmonella typhi, and Staphylococcusi aureus relative to Tetracycline (standard reference drug) [49].
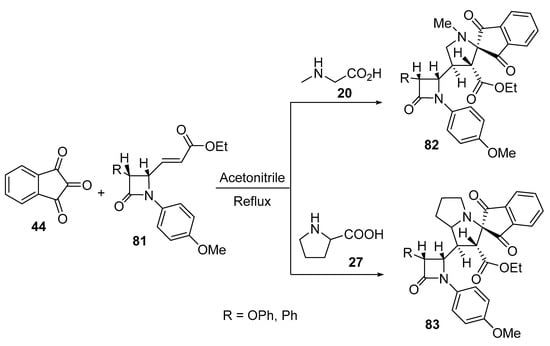
Scheme 26. Synthesis of spiroindanopyrrolidines 82 and spiroindanopyrrolizines 83.
Cycloaddition of cinnamaldehydes 84 with azomethine ylides, generated from another cinnamaldehyde molecule 84 and L-proline 27, afforded hexahydro-1H-pyrrolizines 85 and 86 in different ratios depending on the heating method (conventional heating, 25–80 °C vs. with microwave technique) and the solvent used (MeCN, DMF, toluene, CH2Cl2, DMSO) (Scheme 27) [50].
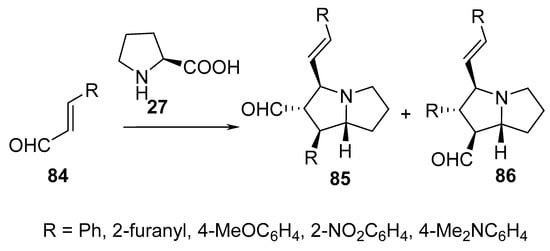
Scheme 27. Synthesis of hexahydro-1H-pyrrolizines 85 and 86.
Pyrrolizidines of type 88 were obtained by reacting β,γ-unsaturated α-keto esters of type 87 with proline 27 in a 2:1 molar ratio. The reaction was assumed to proceed via the formation of azomethine ylides by the condensation of the starting unsaturated esters of type 87 with amino acid 27, which, in turn, interacted with another molecule of 87 to ultimately yield pyrrolizidines of type 88 (Scheme 28) [51].
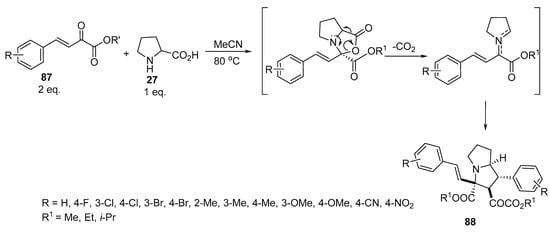
Scheme 28. Synthesis of pyrrolizidines 88.
5. Acrylates
The reaction of O-acryloylacridinediones 89 with azomethine ylides, generated from isatin 19 and secondary amino acids (sarcosine 20/proline 27), afforded the corresponding spiro-pyrrolidines 90 and spiro-pyrrolizidines 91 (Scheme 29) [52].
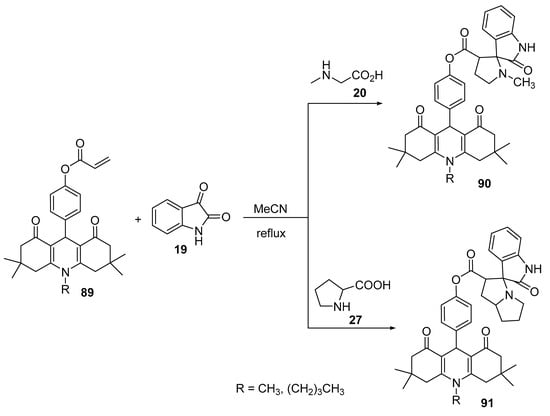
Scheme 29. Synthesis of spiro-pyrrolidines/pyrrolizidines 90/91.
Spiropyrrolidines 94–97 were obtained via the reaction of methyl 2-(1H-inden-2-yl)acrylate 92 with azomethine ylides generated in situ by reacting ketones (isatin 19, acenaphthenequinone 25, ninhydrin 44, or 11H-indeno[1,2-b]quinoxaline-11-one 93) with sarcosine 20 (Scheme 30) [53].
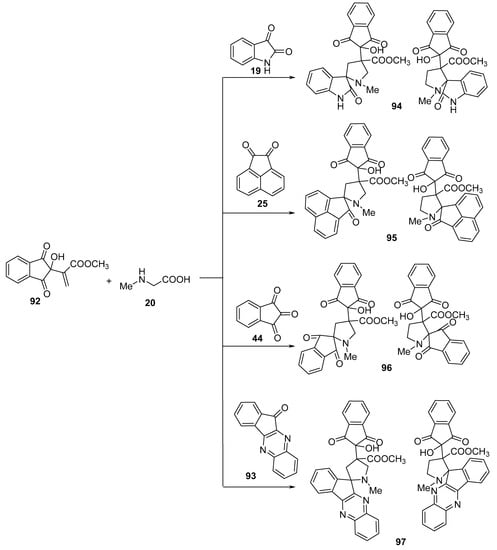
Scheme 30. Synthesis of spiropyrrolidines 94–97.
The reaction of methyl lactate acrylates of type 98 with azomethine ylides, generated from imino-esters 5 in the presence of silver acetate and KOH, gave chiral proline derivatives of type 99 (Scheme 31) [54].
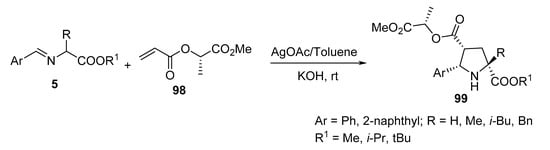
Scheme 31. Synthesis of chiral prolines 99.
The reaction of trans arylacrylates 100 with the azomethine ylide, formed from benzyl-(methoxymethyl)[(trimethylsilyl)methyl]amine 1 in the presence of a catalytic amount of trifluoroacetic acid, afforded the corresponding trans pyrrolidine derivatives 101 (Scheme 32) [55].
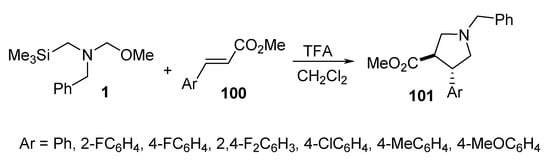
Scheme 32. Synthesis of trans pyrrolidines 101.
6. Intramolecular Cycloaddition Reaction of Azomethine Ylides with Acyclic Unsaturated 2π-Electron Components
6.1. Acyclicunsaturated 2π-Electron Components Containing Olefinic and Aldehyde Groups
Azomethine ylides (formed via the reaction of α-amino esters 103 with O-allyl-5-phenyldiazenylsalicylaldehyde 102) underwent intramolecular [3+2]-cycloaddition under microwave conditions, affording the 8-phenyldiazenylchromeno[4,3-b]pyrrolidines 104 (Scheme 33). The synthesized compounds showed antibacterial activity against Gram-positive (Streptococcus pneumoniae, Clostridium tetani, and Bacillus subtilis) and Gram-negative bacteria (Salmonella typhi, Vibrio cholerae, and Escherichia coli), fungi (Aspergillus fumigatus and Candida albicans), and mycobacteria (M. Tuberculosis H37RV) relative to the antibacterial (Ampicillin, Norfloxacin, Chloramphenicol, Ciprofloxacin), antifungal (Griseofulvin, Nystatin), and antimycobacterial (Metronidazole) standard references used [56].
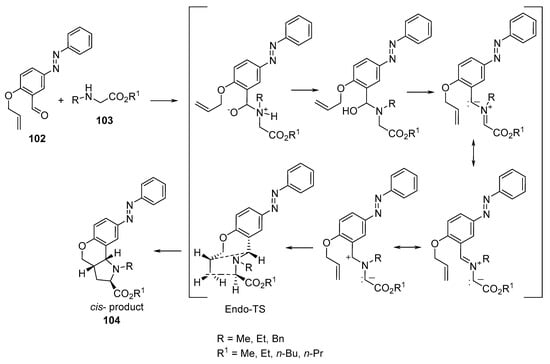
Scheme 33. Synthesis of 8-phenyldiazenylchromeno[4,3-b]pyrrolidines 104.
The intramolecular cycloaddition reaction of azomethine ylides, formed from alkenyl aldehyde 105 and secondary amino acids (sarcosine 20, L-proline 27, thioproline 22, and tetrahydroisoquinoline-3-carboxylic acid 106), afforded the corresponding chromenopyrrole derivatives 107–109 (Scheme 34). The synthesized compounds showed promising antibacterial (against S. aureus, B. subtilis “Gram-positive”; S. pneumoniae, E. coli, and Shigella sp., S. typhi “Gram-negative”) and antifungal (against Trichoderma sp., Aspergillus sp. and C. albicans) activities against the references Tetracycline and Carbendazim (antibacterial and antifungal standard references, respectively) [57].
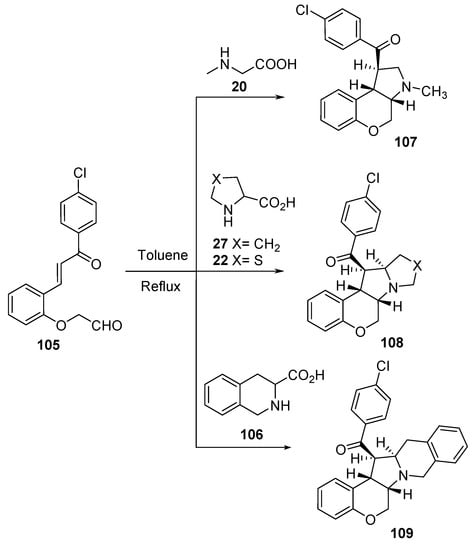
Scheme 34. Synthesis of chromenopyrrole-containing compounds 107–109.
The intramolecular cycloaddition of O-allyl salicylaldehydes 110 and sarcosine 20 under ultrasonic irradiation in methanol at room temperature yielded the corresponding chromeno[4,3-b]pyrroles 111 (Scheme 35) [58].
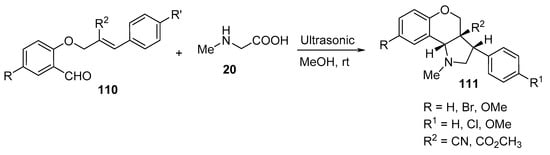
Scheme 35. Synthesis of chromeno[4,3-b]pyrroles 111.
Chromeno[4,3-b]pyrrolidines 113 were obtained in a highly regio- and stereoselective manner by the intramolecular cycloaddition of O-allylic salicylaldehydes 112 and sarcosine 20 (Scheme 36) [59].
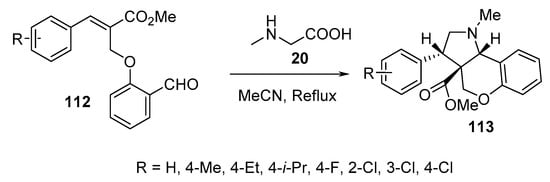
Scheme 36. Synthesis of chromeno[4,3-b]pyrrolidines 113.
Similarly, hexahydrochromeno[4,3-b]pyrroles 116 were obtained via intramolecular [3+2}-cycloaddition of O-allylic salicylaldehyde 114 and amines 115 under microwave conditions (Scheme 37) [60].
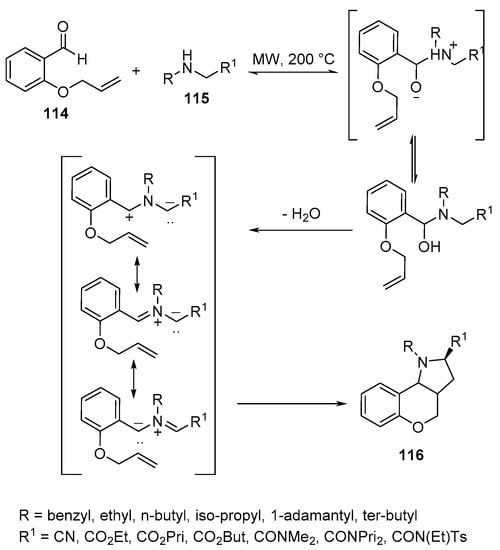
Scheme 37. Synthesis of hexahydrochromeno[4,3-b]pyrroles 116.
Bicyclic pyrrolo[3,4-b]pyrroles 118 were obtained by the intramolecular cyclization of the generated azomethine ylides from aldehydes 117 and sarcosine 20 under refluxing conditions in toluene (Scheme 38) [61].
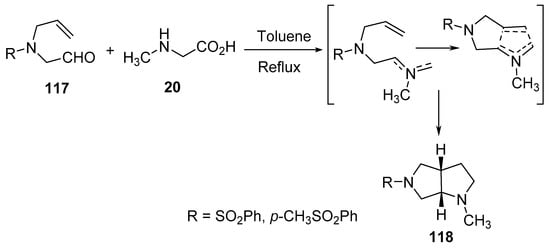
Scheme 38. Synthesis of pyrrolo[3,4-b]pyrroles 118.
Octahydropyrrolo[3,4-b]pyrroles 121 with various substituents in their aromatic rings were synthesized by the intramolecular cycloaddition of azomethine ylides, which was formed from the reaction of alkenyl aldehyde 119 with N-aryl glycines 120 (Scheme 39) [62].
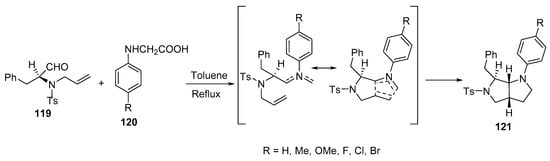
Scheme 39. Synthesis of octahydropyrrolo[3,4-b]pyrroles 121.
The condensation of N-alkenyl aldehydes 122 with α-amino acids (sarcosine 20, thioproline 22 and proline 27) generated azomethine ylides, which underwent an intramolecular cycloaddition reaction yielding the corresponding polycyclic compounds 123 and 124 (Scheme 40) [63].
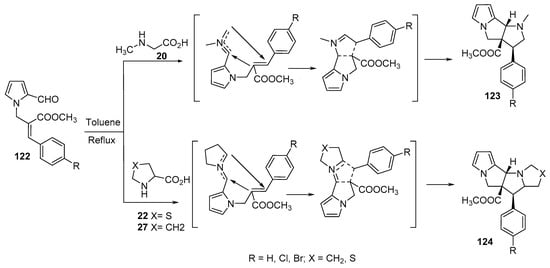
Scheme 40. Synthesis of polycyclic compounds 123 and 124.
Similarly, the intramolecular reaction of azomethine ylide obtained from 2-butenylindole-3-carboxaldehyde 125 with N-methyl glycine ethyl ester hydrochloride 126 gave the indole-containing alkaloid 127. Whereas its reaction with N-methyl glycine 20 or N-allyl glycine 128 gave the corresponding indole heterocycles of type 129 (Scheme 41) [64].
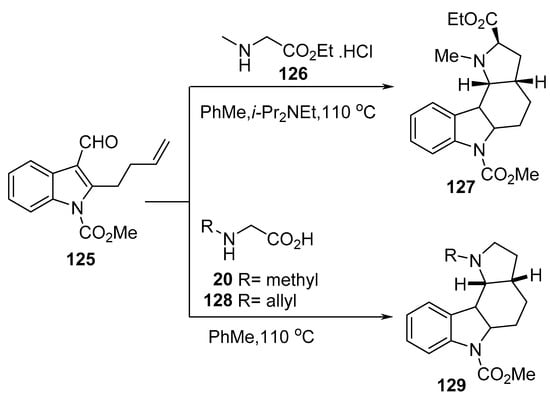
Scheme 41. Synthesis of indole-containing heterocycles 127 and 129.
Another example of intramolecular cycloaddition was the reaction of (E)-2-{[allyl(benzyl)amino]methyl}cinnamaldehydes 130 with proline methyl ester hydrochloride 131 under microwave conditions, which afforded the pyrido[3,4-b]pyrrolizines 132 (Scheme 42) [65].
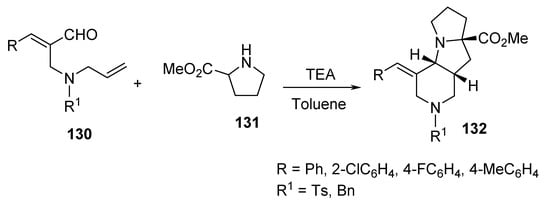
Scheme 42. Synthesis of pyrido[3,4-b]pyrrolizines 132.
By using 1,2-O-cyclohexylidine-3-O-allyl-α-D-xylopentadialdo-1,4-furanose 133 (sugar-derived aldehyde) in a reaction with sarcosine 20, furopyranopyrrolidine of type 134 was formed with high diastereoselectivity (Scheme 43) [66].
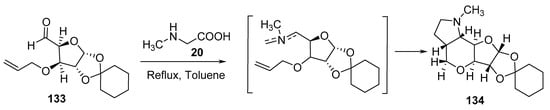
Scheme 43. Synthesis of furopyranopyrrolidine 134.
The intramolecular [3+2]-cycloaddition of azomethine ylides, generated from 2-formylphenyl-(E)-2-phenylethenesulfonates 135 and sarcosine 20, afforded the corresponding [1,2]oxathiino[4,3-b]pyrroles 136. However, the reaction of derivative 135 with L-proline 27 gave the corresponding [1,2]oxathiino[3,4-b]pyrrolizines 137 as trans–trans (major) and cis–trans (minor) isomers (Scheme 44) [67].
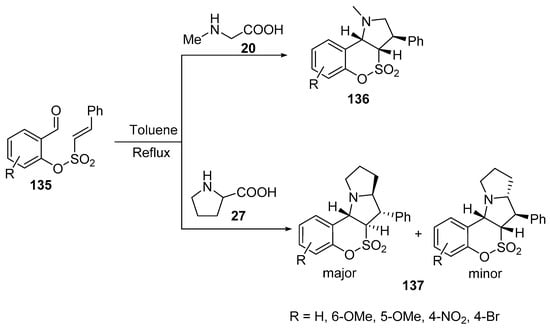
Scheme 44. Synthesis of benzo[e][1,2]oxathiino[4,3-b]pyrrole-4,4-dioxides 136 and benzo[e][1,2]oxathiino[3,4-b]pyrrolizine-6,6-dioxides 137.
Scheme 45 shows an interesting example of a macrocycle of type 139 formation via the intramolecular cycloaddition of an azomethine ylide generated from a triazole-linked glycol-nitroalkenyl aldehyde derivative 138 and sarcosine 20 [68].
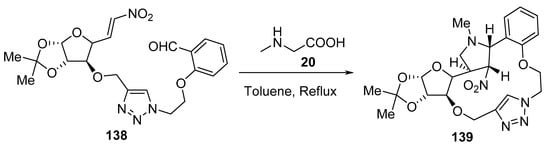
Scheme 45. Synthesis of macrocycle 139.
Polycyclic naphtho[2,1-b]pyrano-pyrrolizidine and indolizidine derivatives 141 and 143 were synthesized by the intramolecular [3+2]-cycloaddition of azomethine ylides generated from naphtho-O-alkenyl aldehydes 140 and α-amino acids (L-proline 27 or DL-pipecolinic acid 142) (Scheme 46) [69].
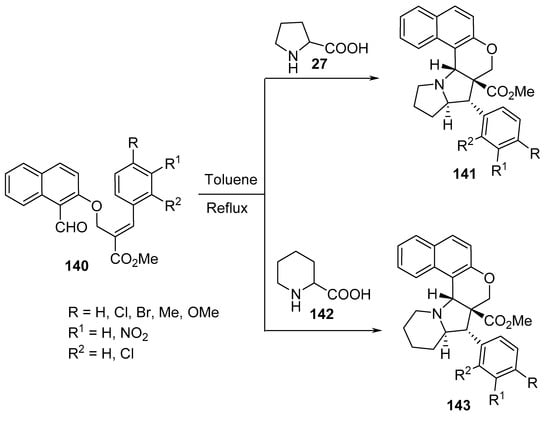
Scheme 46. Synthesis of naptho-pyrano-pyrrolizidines/indolizidines 141 and 143.
6.2. Acyclic Unsaturated 2π-Electron Components Containing Olefinic Linkage and Azirdine
Scheme 47 shows the thermolysis of aziridines 144 that led to the in situ formation of azomethine ylides, which underwent intramolecular cycloaddition, thus affording N-phthalimidopyrrolidine derivatives 145 as a mixture of two diastereoisomers [70].
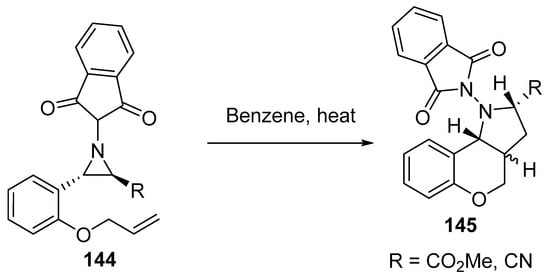
Scheme 47. Synthesis of N-phthalimidopyrrolidines 145.
Another bicyclic system of γ-lactone 147 was created by the intramolecular [3+2]-cycloaddition of azomethine ylide generated via the thermolysis of aziridine derivative 146 in refluxing toluene (Scheme 48) [71].
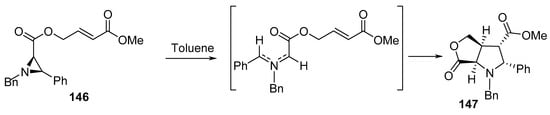
Scheme 48. Synthesis of bicyclic γ-lactone 147.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28020668
References
- Huisgen, R. 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1984; Volume 1, Chapter 1; pp. 3–5.
- Rios-Gutierrez, M.; Domingo, L.R. Unravelling the mysteries of the cycloaddition reactions. Eur. J. Org. Chem. 2019, 2019, 267–282.
- William Lown, J. 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1984; Volume 1, Chapter 6; pp. 657, 726.
- Coldham, I.; Hufton, R. Intramolecular Dipolar Cycloaddition Reactions of Azomethine Ylides. Chem. Rev. 2005, 105, 2765–2809.
- Song, G.; Chen, D.; Su, Y.; Han, K.; Pan, C.-L.; Jia, A.; Li, X. Isolation of Azomethine Ylides and Their Complexes: Iridium(III)-Mediated Cyclization of Nitrone Substrates Containing Alkynes. Angew. Chem. Int. Ed. 2011, 50, 7791–7796.
- Lee, D.J.; Han, H.S.; Shin, J.; Yoo, E.J. Multicomponent Cycloaddition Reaction for the Synthesis of 1,4-Diazepines: Isolation and Reactivity of Azomethine Ylides. J. Am. Chem. Soc. 2014, 136, 11606–11609.
- Molteni, G.; Silvani, A. Spiro-2-oxindoles via 1,3-dipolar cycloadditions. A decade update. Eur. J. Org. Chem. 2021, 2021, 1653–1675.
- Adrio, J.; Carretero, J.C. Stereochemical diversity in pyrrolidine synthesis by catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Chem. Comm. 2019, 55, 11979–11991.
- Bdiri, B.; Zhao, B.-J.; Zhou, Z.-M. Recent advances in the enantioselective 1,3-dipolar cycloaddition of azomethine ylides and dipolarophiles. Tetrahedron Asymm. 2017, 28, 876–899.
- Tang, S.; Zhang, X.; Sun, J.; Niu, D.; Chruma, J.J. 2-Azaallyl Anions, 2-Azaallyl Cations, 2-Azaallyl Radicals, and Azomethine Ylides. Chem. Rev. 2018, 118, 10393–10457.
- Dondas, H.A.; de Gracia Retamosa, M.; Sansano, J.M. Current Trends towards the Synthesis of Bioactive Heterocycles and Natural Products Using 1,3-Dipolar Cycloadditions (1,3-DC) with Azomethine Ylides. Synthesis 2017, 49, 2819–2851.
- Meyer, A.G.; Ryan, J.H. 1,3-Dipolar Cycloaddition Reactions of Azomethine Ylides with Carbonyl Dipolarophiles Yielding Oxazolidine Derivatives. Molecules 2016, 21, 935.
- Fang, X.; Wang, C.-J. Catalytic asymmetric construction of spiropyrrolidines via 1,3-dipolar cycloaddition of azomethine ylides. Org. Biomol. Chem. 2018, 16, 2591–2601.
- Arrastia, I.; Arrieta, A.; Cossio, F.P. Application of 1,3-Dipolar Reactions between Azomethine Ylides and Alkenes to the Synthesis of Catalysts and Biologically Active Compounds. Eur. J. Org. Chem. 2018, 2018, 5889–5904.
- Wei, L.; Chang, X.; Wang, C.-J. Catalytic Asymmetric Reactions with N-Metallated Azomethine Ylides. Acc. Chem. Res. 2020, 53, 1084–1100.
- Verma, S.; George, J.; Singh, S.; Pardasani, P.; Pardasani, R. Cycloaddition reactions of thioisatin with thiazolidine-2-carboxylic acid: A versatile route to new heterocyclic scaffolds. Org. Med. Chem. Lett. 2011, 1, 6.
- Lashgari, N.; Ziarani, G.M. Synthesis of heterocyclic compounds based on isatin through 1,3-dipolar cycloaddition reactions. ARKIVOC 2012, 2012, 277–320.
- Padwa, A.; Pearson, W.H. (Eds.) The chemistry of heterocyclic compounds. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products; John Wiley & Sons: Hoboken, NJ, USA, 2002; Volume 59.
- Martina, K.; Tagliapietra, S.; Veselov, V.V.; Cravotto, G. Green protocols in heterocycle syntheses via 1,3-dipolar cycloadditions. Front. Chem. 2019, 7, 95.
- Grafton, M.; Mansfield, A.C.; Fray, M.J. Dipolar cycloadditions of an unstabilised azomethine ylide under continuous flow conditions. Tetrahedron Lett. 2010, 51, 1026–1029.
- Boruah, M.; Konwar, D.; Sharma, S.D. KF/Al2O3 mediated 1,3-dipolar cycloaddition of azomethine ylides: A novel and convenient procedure for the synthesis of highly substituted pyrrolidines. Tetrahedron Lett. 2007, 48, 4535–4537.
- Belfaitah, A.; Isly, M.; Carboni, B. 1,3-Dipolar cycloadditions of azomethine ylides to alkenylboronic esters. Access to substituted boron analogues of β-proline and 3-hydroxypyrrolidines. Tetrahedron Lett. 2004, 45, 1969–1972.
- Han, Y.; Hou, H.; Fu, Q.; Yan, C.-G. One-pot two-step tandem reactions for selective synthesis of pyrroloisoquinolines and dihydro-, tetrahydro-derivatives. Tetrahedron 2011, 67, 2313–2322.
- Poomathi, N.; Mayakrishnan, S.; Muralidharan, D.; Perumal, P.T. A facile access to novel spiroxindole fused pyrrolidine and thiazolo pyrrolidine benzimidazole derivatives via 1,3-dipolar cycloaddition reaction. Tetrahedron Lett. 2015, 56, 721–726.
- Dandia, A.; Jain, A.K.; Laxkar, A.K.; Bhati, D.S. A highly efficient protocol for the regio- and stereo-selective synthesis of spiro pyrrolidine and pyrrolizidine derivatives by multicomponent reaction. Tetrahedron Lett. 2013, 54, 3180–3184.
- Aksenov, A.V.; Aksenov, D.A.; Arutiunov, N.A.; Aksenov, N.A.; Aleksandrova, E.V.; Zhao, Z.; Du, L.; Kornienko, A.; Rubin, M. Synthesis of Spiro with Anticancer Activity via a Formal -Spirocyclization of Nitroalkenes to Indoles. J. Org. Chem. 2019, 84, 7123–7137.
- Żmigrodzka, M.; Sadowski, M.; Kras, J.; Dresler, E.; Demchuk, O.M.; Kula, K. Polar cycloaddition between N-methyl azomethine ylide and trans-3, 3, 3-trichloro-1-nitroprop-1-ene. Sci. Radices 2022, 1, 26–35.
- Żmigrodzka, M.; Dresler, E.; Hordyjewicz-Baran, Z.; Kulesza, R.; Jasiński, R. Synthesis and chemical properties of 3-alkoxycarbonylchromones and 3-alkoxalylchromones. Chem. Heter. Comp. 2017, 53, 1161–1162.
- Seki, M.; Tsuruta, O.; Tatsumi, R.; Soejima, A. Synthesis and biological evaluation of pyrrolidine derivatives as novel and potent sodium channel blockers for the treatment of ischemic stroke. Bioorg. Med. Chem. Lett. 2013, 23, 4230–4234.
- Sun, H.; Wang, X.; Chen, Y.; Ouyang, L.; Liu, J.; Zhang, Y. Efficient construction of highly functionalized endo’-selective spiro via a regioselective 1,3-dipolar cycloaddition reaction between 3-amino oxindoles as azomethine ylide precursors and nitroalkenes. Tetrahedron Lett. 2014, 55, 5434–5438.
- Alimohammadi, K.; Sarrafi, Y.; Tajbakhsh, M.; Yeganegi, S.; Hamzehloueian, M. An experimental and theoretical investigation of the regio- and stereoselectivity of the polar cycloaddition of azomethine ylides to nitrostyrenes. Tetrahedron 2011, 67, 1589–1597.
- De Silva, N.H.; Pyreddy, S.; Blanch, E.W.; Hügel, H.M.; Maniam, S. Microwave-assisted rapid synthesis of spirooxindole-pyrrolizidine analogues and their activity as anti-amyloidogenic agents. Bioorg. Chem. 2021, 114, 105128.
- Kumar, R.S.; Almansour, A.I.; Arumugam, N.; Mohammad, F.; Kotresha, D.; Menéndez, J.C. Spirooxindole-pyrrolidine heterocyclic hybrids promotes apoptosis through activation of caspase-3. Bioorg. Med. Chem. 2019, 27, 2487–2498.
- Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Alaqeel, S.I.; Krishna, V.S.; Sriram, D. Anti-tubercular activity of novel class of spiropyrrolidine tethered indenoquinoxaline heterocyclic hybrids. Bioorg. Chem. 2020, 99, 103799.
- Almansour, A.I.; Arumugam, N.; Kumar, R.S.; Kotresha, D.; Manohar, T.S.; Venketesh, S. Design, synthesis and cholinesterase inhibitory activity of novel spiropyrrolidine tethered imidazole heterocyclic hybrids. Bioorg. Med. Chem. Lett. 2020, 30, 126789.
- Arumugam, N.; Raghunathan, R. Synthesis of highly functionalized β-lactam substituted pyrroloisoquinoline and indolizinoindole system by sequential intermolecular 1,3-dipolar cycloaddition reaction and Pictet-Spengler cyclization. Tetrahedron 2010, 66, 969–975.
- Wang, H.-T.; Lu, C.-D. Synthesis of 3,4-dihydropyrroloisoquinolines based on cycloaddition initiated by Rh2(cap)4-catalyzed oxidation. Tetrahedron Lett. 2013, 54, 3015–3018.
- Thangamani, A. Regiospecific synthesis and biological evaluation of spirooxindolopyrrolizidines via cycloaddition of azomethine ylide. Eur. J. Med. Chem. 2010, 45, 6120–6126.
- Kaur, A.; Singh, B.; Vyas, B.; Silakari, O. Synthesis and biological activity of 4-aryl-3-benzoyl-5-phenylspiro-2’-one derivatives as novel potent inhibitors of advanced glycation end product. Eur. J. Med. Chem. 2014, 79, 282–289.
- Taghizadeh, M.J.; Arvinnezhad, H.; Samadi, S.; Jadidi, K.; Javidan, A.; Notash, B. Synthesis of new enantiomerically pure spirooxindolopyrrolizidines via a three-component asymmetric 1,3-dipolar cycloaddition reaction of azomethine ylides derived from isatin. Tetrahedron Lett. 2012, 53, 5148–5150.
- Arun, Y.; Saranraj, K.; Balachandran, C.; Perumal, P.T. Novel spirooxindole–pyrrolidine compounds: Synthesis, anticancer and molecular docking studies. Eur. J. Med. Chem. 2014, 74, 50–64.
- Arun, Y.; Bhaskar, G.; Balachandran, C.; Ignacimuthu, S.; Perumal, P.T. Facile one-pot synthesis of novel dispirooxindole-pyrrolidine derivatives and their antimicrobial and anticancer activity against A549 human lung adenocarcinoma cancer cell line. Bioorg. Med. Chem. Lett. 2013, 23, 1839–1845.
- Lakshmi, N.V.; Thirumurugan, P.; Perumal, P.T. An expedient approach for the synthesis of dispiropyrrolidine bisoxindoles, spiropyrrolidine oxindoles and spiroindane-1,3-diones through 1,3-dipolar cycloaddition reactions. Tetrahedron Lett. 2010, 51, 1064–1068.
- Kathirvelan, D.; Haribabu, J.; Reddy, B.S.R.; Balachandran, C.; Duraipandiyan, V. Facile and diastereoselective synthesis of 3,2’-spiropyrrolidineoxindoles derivatives, their molecular docking and antiproliferative activities. Bioorg. Med. Chem. Lett. 2015, 25, 389–399.
- Yuvaraj, P.; Reddy, B.S.R. Synthesis of 3-spiropyrrolidine-3-spirooxindoles from Baylis–Hillman adducts of chromone with azomethine ylides via cycloaddition reaction. Tetrahedron Lett. 2013, 54, 821–827.
- Sarrafi, Y.; Hamzehlouian, M.; Alimohammadi, K.; Khavasi, H.R. Regioselective synthesis of novel spiroindane-1,3-diones through 1,3-dipolar cycloaddition reactions. Tetrahedron Lett. 2010, 51, 4734–4737.
- Kia, Y.; Osman, H.; Kumar, R.S.; Murugaiyah, V.; Basiri, A.; Perumal, S.; Razak, I.A. A facile chemo-, regio- and stereoselective synthesis and cholinesterase inhibitory activity of spirooxindole-pyrrolizine-piperidine hybrids. Bioorg. Med. Chem. Lett. 2013, 23, 2979–2983.
- Kia, Y.; Osman, H.; Kumar, R.S.; Murugaiyah, V.; Basiri, A.; Perumal, S.; Wahab, H.A.; Bing, C.S. Synthesis and discovery of novel piperidone-grafted mono- and bis-spirooxindole-hexahydropyrrolizines as potent cholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 1696–1707.
- Arumugam, N.; Periyasami, G.; Raghunathan, R.; Kamalraj, S.; Muthumary, J. Synthesis and antimicrobial activity of highly functionalised novel β-lactam grafted spiropyrrolidines and pyrrolizidines. Eur. J. Med. Chem. 2011, 46, 600–607.
- Hong, B.-C.; Liu, K.-L.; Tsai, C.-W.; Liao, J.-H. Proline-mediated dimerization of cinnamaldehydes via 1,3-dipolar cycloaddition reaction with azomethine ylides. A rapid access to highly functionalized hexahydro-1H-pyrrolizine. Tetrahedron Lett. 2008, 49, 5480–5483.
- Kang, T.-R.; Cheng, Y.; He, L.; Ye, J.; Liu, Q.-Z. Facile synthesis of highly functional pyrrolizidine derivatives from β,γ-unsaturated α-keto esters and proline via a tandem cycloaddition. Tetrahedron Lett. 2012, 53, 2552–2555.
- Rajesh, R.; Suresh, M.; Selvam, R.; Raghunathan, R. Synthesis of acridinedione derived mono spiro-pyrrolidine/pyrrolizidine derivatives–a facile approach via intermolecular cycloaddition reaction. Tetrahedron Lett. 2014, 55, 4047–4053.
- Ramesh, E.; Kathiresan, M.; Raghunathan, R. Solvent-free microwave-assisted conversion of Baylis–Hillman adducts of ninhydrin into functionalized spiropyrrolidines/pyrrolizidines through 1,3-dipolar cycloaddition. Tetrahedron Lett. 2007, 48, 1835–1839.
- Nájera, C.; de Gracia Retamosa, M.; Sansano, J.M. 1,3-Dipolar cycloadditions of azomethine ylides with chiral acrylates derived from methyl (S)- and (R)-lactate: Diastereo- and enantioselective synthesis of polysubstituted prolines. Tetrahedron Asymmetry 2006, 17, 1985–1989.
- Ujjainwalla, F.; Warner, D.; Snedden, C.; Grisson, R.D.; Walsh, T.F.; Wyvratt, M.J.; Kalyani, R.N.; MacNeil, T.; Tang, R.; Weinberg, D.H.; et al. Design and syntheses of melanocortin subtype-4 receptor agonists. Part 2: Discovery of the dihydropyridazinone motif. Bioorg. Med. Chem. Lett. 2005, 15, 4023–4028.
- Parmar, N.J.; Pansuriya, B.R.; Barad, H.A.; Kant, R.; Gupta, V.K. An improved microwave assisted one-pot synthesis, and biological investigations of some novel aryldiazenyl chromeno fused pyrrolidines. Bioorg. Med. Chem. Lett. 2012, 22, 4075–4079.
- Purushothaman, S.; Prasanna, R.; Niranjana, P.; Raghunathan, R.; Nagaraj, S.; Rengasamy, R. Stereoselective synthesis of hexahydro-3-methyl-1-arylchromenopyrrole and its annulated heterocycles as potent antimicrobial agents for human pathogens. Bioorg. Med. Chem. Lett. 2010, 20, 7288–7291.
- Ramesh, E.; Raghunathan, R. A facile synthesis of chromenopyrroles derived from allyl derivatives of Baylis–Hillman adducts through intramolecular 1,3-dipolar cycloaddition using ultrasonication. Tetrahedron Lett. 2008, 49, 1125–1128.
- Bakthadoss, M.; Sivakumar, N.; Sivakumar, G.; Murugan, G. Highly regio- and stereoselective synthesis of tricyclic frameworks using Baylis–Hillman derivatives. Tetrahedron Lett. 2008, 49, 820–823.
- Pospíšil, J.; Potáček, M. Microwave-assisted solvent-free intramolecular 1,3-dipolar cycloaddition reactions leading to hexahydrochromenopyrroles: Scope and limitations. Tetrahedron 2007, 63, 337–346.
- Poornachandran, M.; Raghunathan, R. Synthesis of pyrrolopyrroles and perhydrothiazolopyrrolopyrroles. Tetrahedron 2008, 64, 6461–6474.
- Poornachandran, M.; Raghunathan, R. A novel diastereoselective 1,3-dipolar cycloaddition approach to cis-fused bispyrrolidines. Tetrahedron Asymmetry 2008, 19, 2177–2183.
- Kathiravan, S.; Ramesh, E.; Raghunathan, R. Synthesis of pyrrolopyrrolizine and pyrrolizinepyrrolizine derived from allyl derivatives of Baylis–Hillman adducts through intramolecular 1,3-dipolar cycloaddition. Tetrahedron Lett. 2009, 50, 2389–2391.
- Coldham, I.; Dobson, B.C.; Franklina, A.I.; Fletcher, S.R. Synthesis of tetracyclic indole-containing ring systems by intramolecular cycloadditions of azomethine ylides. Tetrahedron Lett. 2007, 48, 873–875.
- Mishra, A.; Rastogi, N.; Batra, S. 2-(N-Allylaminomethyl)cinnamaldehydes as substrates for syntheses of aza-polycycles via intramolecular cycloaddition reactions. Tetrahedron 2012, 68, 2146–2154.
- Sirisha, N.; Raghunathan, R. Stereoselective synthesis of novel glyco-pyrano pyrrolidines/pyrrolizidines/indolizidines through intramolecular cycloaddition approach. Tetrahedron Lett. 2010, 51, 2515–2518.
- Ghandi, M.; Taheri, A.; Bozcheloei, A.H.; Abbasi, A.; Kia, R. Synthesis of novel tricyclic and tetracyclic sultone scaffolds via intramolecular 1,3-dipolar cycloaddition reactions. Tetrahedron 2012, 68, 3641–3648.
- Rao, J.N.S.; Raghunathan, R. One-pot sequential azide–alkyne/intramolecular azomethine ylide 1,3-dipolar cycloaddition strategy for the synthesis of carbohydrate grafted macrocycles. Tetrahedron Lett. 2015, 56, 2669–2673.
- Kathiravan, S.; Vijayarajan, D.; Raghunathan, R. Novel synthesis of naphthopyrano pyrrolizidines and indolizidines through intramolecular 1,3-dipolar cycloaddition reaction. Tetrahedron Lett. 2010, 51, 3065–3070.
- Pankova, A.S.; Voronin, V.V.; Kuznetsov, M.A. Intramolecular cycloaddition of N-phthalimidoaziridines to double and triple carbon–carbon bonds. Tetrahedron Lett. 2009, 50, 5990–5993.
- Shishido, Y.; Ito, F.; Morita, H.; Ikunaka, M. Stereoselective synthesis of a novel 2-aza-7-oxabicyclooctane as neurokinin-1 receptor antagonist. Bioorg. Med. Chem. Lett. 2007, 17, 6887–6890.
This entry is offline, you can click here to edit this entry!