Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Genome-scale metabolic models (GEMs) aim to systematically encode knowledge of the metabolism of an organism. GEMs are composed of different layers of information and are constructed with a combination of automated approaches and manual curation based on the available literature and experimental data. These models not only encode existing knowledge about an organism, but can also generate new knowledge through various analytical methods. The latter are mostly focused on the assessment of reaction fluxes through the metabolic network in different conditions.
- context-specific genome scale metabolic modelling
- constraint-based modelling
- omics data integration
- metabolism
- genome scale metabolic modelling
- systems biology
- systems medicine
1. Introduction
Genome-scale metabolic models (GEMs) have found numerous applications in different domains, ranging from biotechnology to systems medicine [1]. One of their main benefits is that they can provide genotype-to-phenotype projections, such as growth rate and nutrient uptake predictions, and predictions of metabolic flux values. The latter can be used to assess metabolic reaction activities in different contexts [2,3]. A GEM describes a metabolic network with a stoichiometric matrix [4] and each reaction is constrained by its minimal and maximal flux bounds. Moreover, a GEM usually encodes the information on gene–protein reaction (GPR) associations, which can be applied in the adaptation of a GEM to a specific context described with high-throughput data, such as transcriptomics or proteomics data. Such integration can be performed with the application of context-specific model reconstruction algorithms, which are used to adapt the flux bounds of a reference model to a given context described with (at least one) high-throughput dataset. This allows one to at least partially automatise the reconstruction of tissue-specific, cell type-specific, disease-specific, or even personalised GEMs. Further investigation of context-specific GEMs includes comparative analyses between different conditions (e.g., analysis of metabolic reprogramming in cancer cells [5]), and identification of biomarkers and therapeutic targets in different diseases or disorders [6].
2. Genome-Scale Metabolic Modelling
Genome-scale metabolic models (GEMs) aim to systematically encode our knowledge of the metabolism of an organism. Reference GEMs describing generic models of a cell are constructed with a combination of automated approaches and manual curation. Such reconstructions are based on genome annotation data and a myriad of additional data sources, including biochemical databases, organism-specific databases, experimental data, and literature data [7]. GEM reconstruction, its refinement, adaptation, and analysis are commonly performed with the aid of model building tools [8] and reconstruction and analysis frameworks, such as COBRA [9], COBRApy [10], RAVEN [11] or PSAMM [12]. These frameworks provide implementation of a vast scope of methods with different goals, including the reconstruction of a draft metabolic model [13], visualisation of metabolic maps (e.g., see Paint4Net [14]), identification of blocked reactions and gap filling [15] and analysis of reconstructed GEMs, such as optimal steady-state flux assessment [16] or flux sampling [17]. GEMs have been reconstructed for more than 1,000 different organisms [18]. Moreover, advances in our knowledge guide iterative refinements of GEMs. For example, Recon presents a generic human GEM that has gone through several iterations from Recon 1 [19] to Recon 2.2 [20] and to Recond3D [21], and was later extended and integrated with the HMR2.0 database [22] to obtain the Human–GEM model [23].
In the context of biomedicine, GEM applications range from the identification of disease biomarkers to the prediction of drug targets [24], drug repurposing [25] and cancer research [26]. GEMs can also be applied in a vast array of bioengineering applications [18]. These range from predicting cellular phenotypes (e.g., in the context of predicting maximal growth in different conditions and identification of an optimal medium [27]) to guiding metabolic engineering (e.g., in the context of optimal strain design [28]) and identification of a minimal gene set [29].
Most computational approaches aimed at the analysis of GEMs rely on constraint-based modelling and are based on flux balance analysis (FBA) [16] or its derivations. FBA aims to find the metabolic flux values that are consistent with a set of given constraints (minimal and maximal flux bounds) and which bring the system to a steady state. Moreover, FBA requires a specification of required metabolic functionality (RMF) that is used to define an objective function for optimisation. The optimisation can then be formulated as a linear programming (LP) problem. However, since the constraints in this formulation are usually mathematically underdetermined [30], several nonunique optimal solutions exist. To assess metabolic flux ranges through reactions that bring the system to a near optimal, or optimal, steady state, flux variability (FVA) can be used [31]. However, the latter still requires the specification of a RMF, which is hard to identify in a general context and may yield biased results. Moreover, it has been shown that the definition of the RMF strongly affects the precision of model predictions [32]. An unbiased alternative to methods relying on RMF-based optimisation is to use flux sampling of the feasible solution space without a specific optimisation criterion [17].
Reconstructed GEMs, as described above, present the metabolism of a general cell in an arbitrary context and, thus, compose generic models. Since only specific metabolic reactions are, in fact, active in a specific cell [33], these models need to be further tailored to a specific context in which only a subset of enzymes is active [34]. This process can be carried out using different reconstruction algorithms, in combination with high-throughput datasets and available biological knowledge, to obtain context-specific models (see Figure 1 and Tables 1 and 2). The latter present a subset of a generic GEM and can be used to describe the metabolism of a specific cell in a specific context [35]. Finally, such a model can describe a cell-, a tissue-, a disease-, or even an individual-specific model.
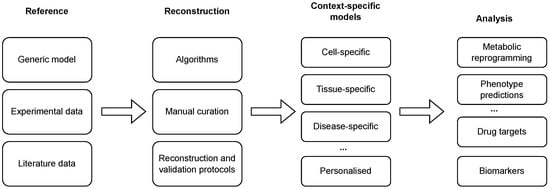
Figure 1. Reconstruction and analysis of context-specific GEMs. Generic models are tailored to a specific context with the integration of (high-throughput) experimental and literature data using a combination of automated algorithms and manual curation. The reconstruction process can be additionally enhanced with the application of reconstruction and validation protocols. The reconstructed models can be used to conduct different analyses, ranging from the prediction of phenotypes and context-specific reprogramming of a metabolic network to the identification of drug targets and disease biomarkers.
3. Algorithms and Tools for Reconstruction of Context-Specific Models
Most algorithms for the reconstruction of context-specific GEMs rely on transcriptomics data to identify active and inactive genes and to adjust metabolic reaction activities in a given context (see Table 2). In this case, each transcript and its corresponding protein/enzyme needs to be associated with specific reactions. One of the first attempts to correlate gene expression data with metabolic flux constraints was presented by Akesson et al. [36]. This was performed on a gene-by-gene basis, where fluxes through the metabolic reactions, with experimental evidence suggesting the absence of their enzymes, were constrained to 0.
Gene–protein reaction (GPR) rules present an association between a specific gene and a metabolic reaction in a model. These rules can describe different types of gene–reaction linkage. For example, different genes might encode different subunits of the same enzyme. In this case, a reaction catalysed by this enzyme can be active only when all of the respective genes are expressed (AND rule). Different genes might also express isoforms of the same enzyme. In this case, a reaction catalysed by this enzyme can be active when at least one of the respective genes is expressed (OR rule) [37]. A large number of recent algorithmic approaches for the reconstruction of context-specific GEMs rely on GPR rules to project the transcriptomics data to reaction activities. However, as illustrated above, GPR rules are encoded in a model as Boolean functions. On the other hand, gene expression data are usually described with non-binary values. In this case, logical OR can be interpreted as the maximum, and logical AND as the minimum, between two or more values (Min/Max GPR mapping) [38]. Alternatively, AND can also be interpreted as the geometric mean, and OR as the sum of two or more values [39].
Certain algorithms only require a definition of a core set of reactions, which are active in a given context. A list of such reactions can be compiled manually (e.g., see [40]) or automatically using transcriptomics data (e.g., see [41,42]). Some reconstruction algorithms allow the integration of other kinds of data, for example metabolomics or proteomics data (see Table 2).
Researchers employ and extend the classification of methods as introduced in [43]. Namely, the majority of the methods can be classified into three main families, i.e., GIMME-, iMAT-, and MBA-like families. The researchers also introduce a MADE-like family, which employs differential expression data in the reconstruction process (see Table 1 and Figure 2). An overview of the algorithms for context-specific GEM reconstruction is summarised in Table 2.
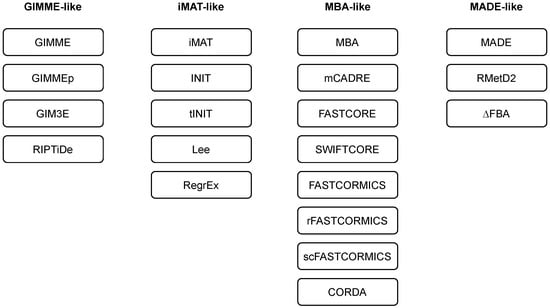
Figure 2. Families of algorithms for automated reconstruction of context-specific models.
Table 1. An overview of different families of algorithms for context-specific model reconstruction. Abbreviations: RMF—required metabolic function; MILP–mixed integer linear programming.
| Family | Description |
|---|---|
| GIMME-like | Maximising the compliance with the experimental evidence while pertaining to a given RMF. |
| iMAT-like | Does not specify a RMF, matching of reactions states (active or inactive) with expression profiles (present or absent), employs MILP-based optimisation. |
| MBA-like | Defining core reactions and removing other reactions while pertaining to model consistency, support integration of different data types. |
| MADE-like | Employs differential gene expression data to identify flux differences between two or more conditions. |
Table 2. An overview of algorithms for automated reconstruction of context-specific models. Abbreviations: LP—linear programming; RMF—required metabolic function.
| Algorithm | Reference | Family | Input Data | Comments |
|---|---|---|---|---|
| GIMME | Becker et al., 2008 [38] | GIMME-like | transcriptomics | Inactivate reactions below a threshold while maintaining RMF. |
| GIMMEp | Bordbar et al., 2012 [44] | GIMME-like | transcriptomics, proteomics | RMFs based on proteomics data. |
| GIM3E | Schmidt et al., 2013 [45] | GIMME-like | transcriptomics, metabolomics | No thresholding. |
| RIPTiDe | Jenior et al., 2020 [46] | GIMME-like | transcriptomics | Minimises the weighted flux values, no thresholding. |
| iMAT | Zur et al., 2010 [47] | iMAT-like | transcriptomics, proteomics | Matches reaction activities with expression profiles, no RMF. |
| INIT | Agren et al., 2012 [48] | iMAT-like | transcriptomics, proteomics, metabolomics (qualitative) | Reaction weights based on experimental evidence, integration of metabolomics data. |
| tINIT | Agren et al., 2014 [49] | iMAT-like | prior knowledge, transcriptomics, proteomics, metabolomics (qualitative) | Based on a set of required metabolic tasks. |
| Lee | Lee et al., 2012 [50] | iMAT-like | transcriptomics | Uses absolute expression data (RNA-seq). |
| RegrEx | Estevez et al., 2015 [51] | iMAT-like | transcriptomics | Uses absolute expression data (RNA-seq) and regularisation. |
| MBA | Jerby et al., 2010 [52] | MBA-like | prior knowledge, transcriptomics, proteomics, metabolomics, fluxomics | Removes non-core reactions and checks model consistency for core reactions. |
| mCADRE | Wang et al., 2012 [53] | MBA-like | transcriptomics, metabolomics | Different reaction scores to determine core reactions. |
| FASTCORE | Vlassis et al., 2014 [40] | MBA-like | a set of core reactions | Two LPs to find a minimal set of non-core reactions to activate all core reactions. |
| SWIFTCORE | Tefagh and Boyd, 2020 [54] | MBA-like | a set of core reactions | Enhanced runtime and network compactness in comparison to FASTCORE. |
| FASTCORMICS | Pires Pacheco at al., 2015 [41] | MBA-like | transcriptomics | FASTCORE workflow for microarray data. |
| rFASTCORMICS | Pires Pacheco at al., 2019 [42] | MBA-like | transcriptomics | FASTCORE workflow for RNA-seq data. |
| scFASTCORMICS | Pires Pacheco at al., 2022 [55] | MBA-like | transcriptomics | FASTCORE workflow for scRNA-seq data. |
| CORDA | Schultz and Qutub, 2016 [34] | MBA-like | a set of core reactions | Does not require to remove all non-core reactions. |
| MADE | Jensen and Papin, 2011 [56] | MADE-like | transcriptomics | Identifies reaction activities in a sequence of measurements. |
| RMetD2 | Zhang et al., 2019 [57] | MADE-like | transcriptomics | Sequentially pushes the constraints. |
| ΔFBA | Ravi et al., 2021 [58] | MADE-like | transcriptomics | Finds a consistent and minimal solution of flux differences between the conditions. |
This entry is adapted from the peer-reviewed paper 10.3390/metabo13010126
This entry is offline, you can click here to edit this entry!