Cancer ranks as a leading cause of death and a significant barrier to increasing life expectancy in every country. According to estimates from the World Health Organization (WHO) in 2019, cancer is the first or second leading cause of death before age 70 in 112 of 183 countries and ranks third or fourth in 23 countries. Tumor treatments usually consist of chemotherapy, radiotherapy, surgery, targeted therapy, immunotherapy, and endocrine therapy that can be used individually or in combination, depending on the stage and type of tumor and diagnosis. Several factors reduce the practical improvement of a cancer patient’s prognosis. Drug-untreatable targets, chemoresistance, tumor heterogeneity, and metastases formation are significant barriers to the effective cure of cancer patients. Despite intense preclinical and clinical research efforts, the survival rate of patients suffering from the most aggressive cancer types has not yet improved, mainly due to therapeutic failure. Moreover, nearly all anticancer medications on the market have serious adverse effects; therefore, new, safer anticancer drugs are desirable.
- Mebendazole
- drug repurposing
- antitumor therapy
- brain cancer
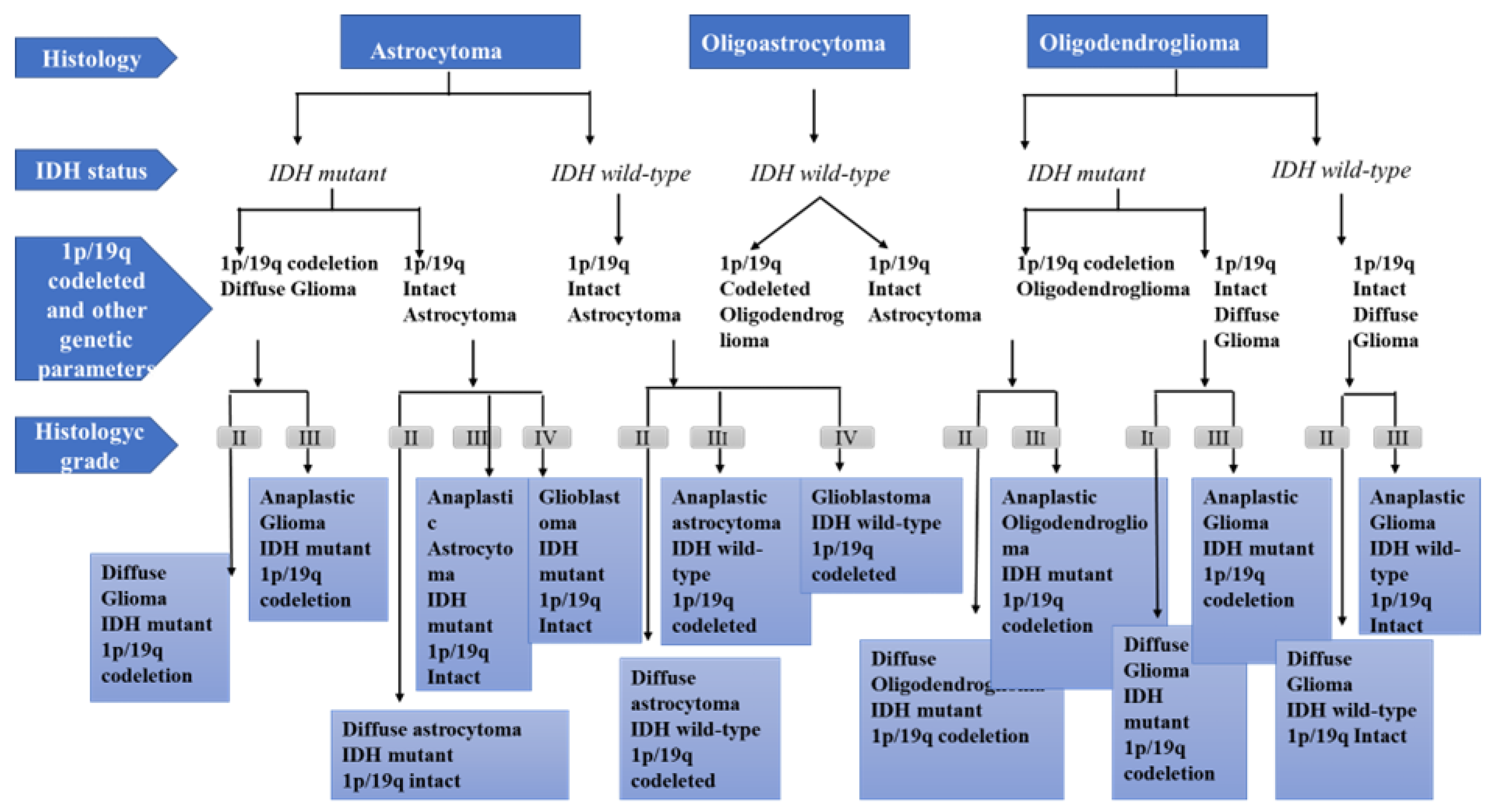
1. Physiopathology of Glioma Development
-
Grade I gliomas are low-grade benign lesions, such as pilocytic astrocytomas, that have limited proliferation and are frequent in children; however, they might acquire higher levels of malignancy. Surgical resection is a good therapeutic option in most cases.
-
Grade II gliomas have low-grade but infiltrative lesions that tend to show higher recurrence after surgical resection.
-
Grade III gliomas have intermediate- to high-grade lesions with atypia, higher mitotic activity, and evidence of malignancy. Patients with these tumors usually are prescribed additional radiation as well as chemotherapy.
-
Grade IV tumors are high-grade malignant lesions with higher mitotic activity, microvascular proliferation, high necrosis, and the worst prognosis.
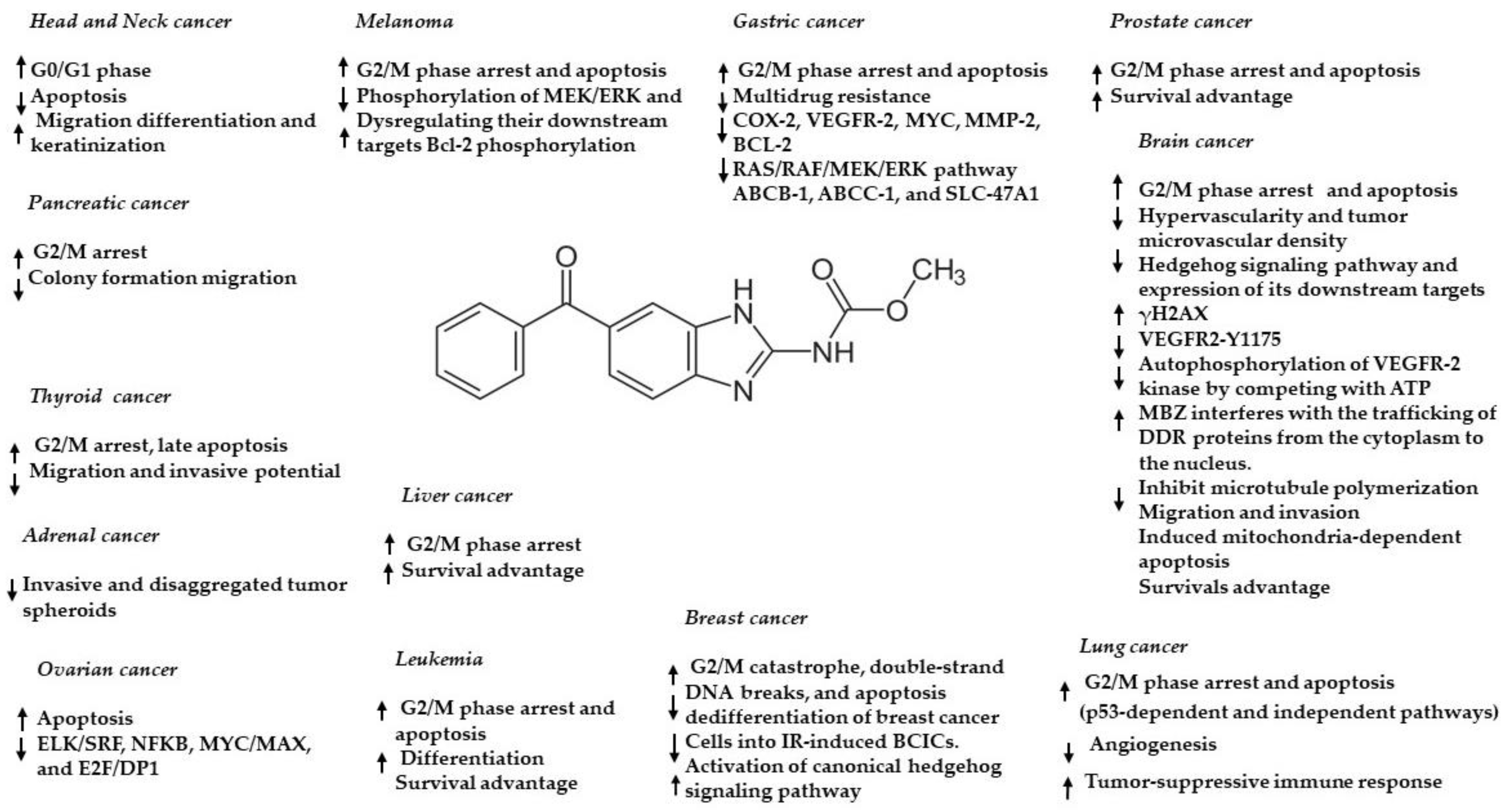
2. Mebendazole: Chemical Structure and Pharmacokinetic Properties
3. Preclinical Evidence of Mebendazole in Brain Cancer—In Vitro and In Vivo
This entry is adapted from the peer-reviewed paper 10.3390/ijms24021334
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. World Health Organization Classification of Tumours of the Central Nervous System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 2, 1231–1251.
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild-type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718.
- Giannini, C.; Burger, P.C.; Berkey, B.A.; Cairncross, J.G.; Jenkins, R.B.; Mehta, M.; Curran, W.J.; Aldape, K. Anaplastic oligodendroglial tumors: Refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol. 2008, 18, 360–369.
- Perez-Larraya, J.G.; Ducray, F.; Chinot, O.; Catry-Thomas, I.; Taillandier, L.; Guillamo, J.-S.; Campello, C.; Monjour, A.; Cartalat-Carel, S.; Barrie, M.; et al. Temozolomide in elderly patients with newly diagnosed glioblastoma and poor performance status: An ANOCEF phase II trial. J. Clin. Oncol. 2011, 29, 3050–3055.
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435.
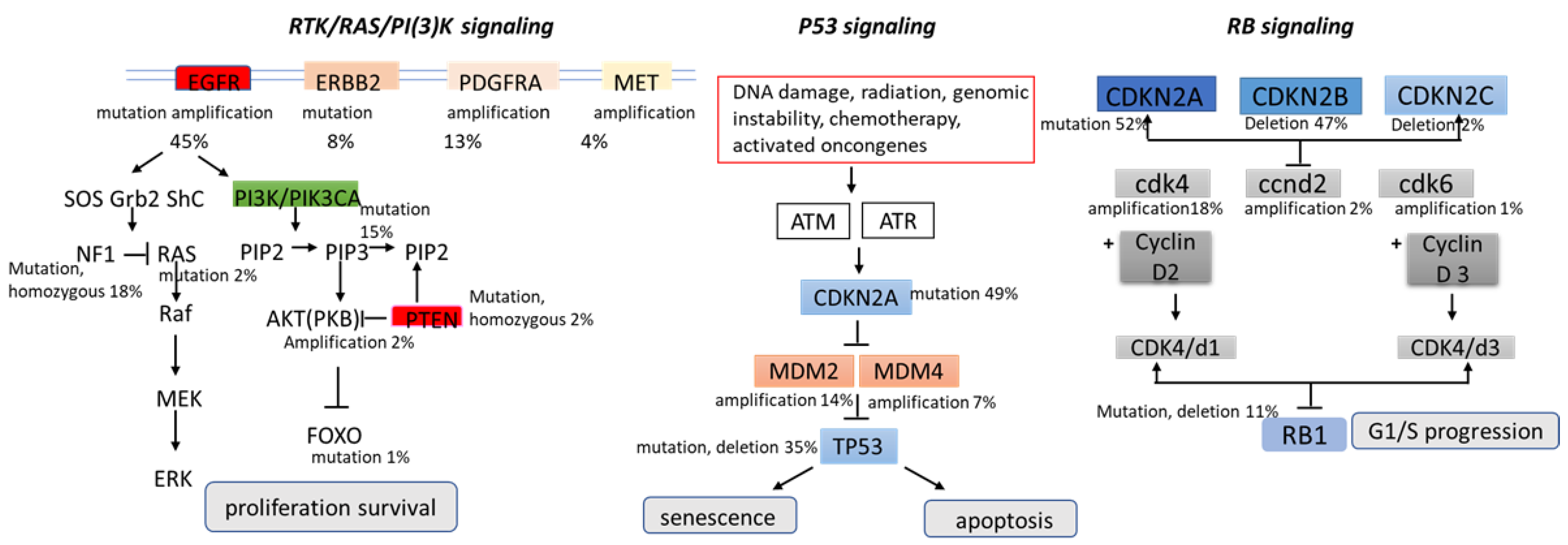
- Wang, H.; Xu, T.; Jiang, Y.; Xu, H.; Yan, Y.; Fu, D.; Chen, J. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia 2015, 17, 239–255.
- Hatanpa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684.
- Nazarenko, I.; Hede, S.M.; He, X.; Hedren, A.; Thompson, J.; Lindstrom, M.S.; Nister, M. PDGF and PDGF receptors in glioma. Upsala J. Med Sci. 2012, 117, 99–112.
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 12, 1831.
- Yip, S.; Iafrate, A.J.; Louis, D.N. Molecular diagnostic testing in malignant gliomas: A practical update on predictive markers. J. Neuropathol. Exp. Neurol. 2008, 67, 1–15.
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606.
- Mao, H.; LeBrun, D.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56.
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265.
- Karsy, M.; Neil, J.A.; Guan, J.; Mahan, M.A.; Colman, H.; Jensen, R.L. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurg. Focus. 2015, 38, E4.
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 19–331.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453.
- Yeu, Y.; Yoon, Y.; Park, S. Protein localization vector propagation: A method for improving the accuracy of drug repositioning. Mol. Biosyst. 2015, 11, 2096–2102.
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299.
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306.
- Brugmans, J.P.; Theinpont, D.C.; Wijngaarden, I.V.; Vanparijs, O.F.; Schuermans, V.L.; Lauwers, H.L. Mebendazole in enterobiasis. Radiochemical and pilot clinical study in 1278 subjects. JAMA 1971, 217, 313–316.
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-mebendazole as an anti-cancer agent. Ecancermedicalscience 2014, 8, 443.
- Reuter, S.; Jensen, B.; Buttenschoen, K.; Kratzer, W.; Kern, P. Benzimidazoles in the treatment of alveolar echinococcosis: A comparative study and review of theliterature. J. Antimicrob. Chemother. 2000, 46, 451–456.
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a candidate for drug repurposing in oncology: An extensive review of current literature. Cancers 2019, 11, 1284.
- Yangco, B.G.; Klein, T.W.; Deresinski, S.C.; Vickery, A.C.; Craig, C.P. Flubendazole andmebendazole in the treatment of trichuriasis and other helminthiases. Clin. Ther. 1981, 4, 285–290.
- Tolomeo, M.; Colomba, C.; Meli, M.; Cascio, A. Hepatotoxicity caused by Mebendazole in a patient with Gilbert’s syndrome. J. Clin. Pharm. Ther. 2019, 44, 985–987.
- Karra, N.; Cohen, R.; Berlin, M.; Dinavitser, N.; Koren, G.; Berkovitch, M. Safety of mebendazole use during lactation: A case series report. Drugs R D 2016, 16, 251–254.
- De Silva, N.; Guyatt, H.; Bundy, D. Anthelmintics. A comparative review of their clinical pharmacology. Drugs 1997, 53, 769–788.
- Göçmen, A.; Toppare, M.F.; Kiper, N. Treatment of hydatid disease in childhood with Mebendazole. Eur. Respir. J. 1993, 6, 253–257.
- Chen, K.T.; Twu, S.J.; Chang, H.J.; Lin, R.S. Outbreak of Stevens-Johnson syndrome/toxic epidermal necrolysis associated with mebendazole and metronidazole use among Filipino laborers in Taiwan. Am. J. Public Health 2003, 93, 489–492.
- Edwards, G.; Brechenridge, A.M. Clinical pharmacokinetics of anthelmintic drugs. Clin. Pharmacokinet. 1988, 15, 67–93.
- Liu, C.-S.; Zhang, H.-B.; Jiang, B.; Yao, J.-M.; Tao, Y.; Xue, J.; Wen, A.-D. Enhanced bioavailability and cysticidal effect of three mebendazole-oil preparations in mice infected with secondary cysts of Echinococcus granulosus. Parasitol. Res. 2012, 111, 1205–1211.
- Alavi, S.E.; Ebrahimi Shahmabadi, H. Anthelmintics for drug repurposing: Opportunities and challenges. Saudi Pharm. J. 2021, 29, 434–445.
- De la Torre-Iglesias, P.M.; García-Rodriguez, J.J.; Torrado, G.; Torrado, S.; Torrado-Santiago, S.; Bolás-Fernández, F. Enhanced bioavailability and anthelmintic efficacy of Mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose. Drug Des. Dev. Ther. 2014, 18, 1467–1479.
- Münst, G.J.; Karlaganis, G.; Bircher, J. Plasma concentrations of Mebendazole during treatment of echinococcosis. Eur. J. Clin. Pharmacol. 1980, 17, 375–378.
- Braithwaite, P.A.; Roberts, M.S.; Allan, R.J.; Watson, T.R. Clinical pharmacokinetics of high dose mebendazole in patients treated for cystic hydatid disease. Eur. J. Clin. Pharmacol. 1982, 22, 161–169.
- Bekhti, A.; Pirotte, J. Cimetidine increases serum mebendazole concentrations. Implications for treatment of hepatic hydatid cysts. Br. J. Clin. Pharmacol. 1987, 24, 390–392.
- Luder, P.J.; Siffert, B.; Witassek, F.; Meister, F.; Bircher, J. Treatment of hydatid disease with high oral doses of Mebendazole. Eur. J. Clin. Pharmacol. 1986, 31, 443–448.
- Corti, N.; Heck, A.; Rentsch, K.; Zingg, W.; Jetter, A.; Stieger, B.; Pauli-Magnus, C. Effect of ritonavir on the pharmacokinetics of the benzimidazoles albendazole and Mebendazole: An interaction study in healthy volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 999–1006.
- Rodriguez-Caabeiro, F.; Criado-Fornelio, A.; Jimenez-Gonzalez, A.; Guzman, L.; Igual, A.; Perez, A.; Pujol, M. Experimental chemotherapy and toxicity in mice of three Mebendazole polymorphic forms. Chemotherapy 1987, 33, 266–271.
- Swanepoel, E.; Liebenberg, W.; de Villiers, M.M. Quality evaluation of generic drugs by dissolution test: Changing the USP dissolution medium to distinguish between active and non-active Mebendazole polymorphs. Eur. J. Pharm. Biopharm. 2003, 55, 345–349.
- Charoenlarp, P.; Waikagul, J.; Muennoo, C.; Srinophakun, S.; Kitayaporn, D. Efficacy of single-dose Mebendazole, polymorphic forms A and C, in the treatment of hookworm and Trichuris infections. Southeast Asian J. Trop. Med. Public Health 1993, 24, 712–716.
- Bai, R.Y.; Staedtke, V.; Wanjiku, T.; Rudek, M.A.; Joshi, A.; Gallia, G.L.; Riggins, G.J. Brain Penetration and Efficacy of Different Mebendazole Polymorphs in a Mouse Brain Tumor Model. Clin. Cancer Res. 2015, 21, 3462–3470.
- Son, D.S.; Lee, E.S.; Adunyah, S.E. The antitumor potentials of benzimidazole anthelmintics as repurposing drugs. Immun. Netw. 2020, 20, 29.
- Armando, R.G.; Mengual Gómez, D.L.; Gomez, D.E. New drugs are not enough drug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684.
- Nath, J.; Paul, R.; Ghosh, S.K.; Paul, J.; Singha, B.; Debnath, N. Drug repurposing andrelabeling for cancer therapy: Emerging benzimidazole antihelminthics with potent anticancer effects. Life Sci. 2020, 1, 118189.
- Williamson, T.; Mendes, T.B.; Joe, N.; Cerutti, J.M.; Riggins, G.J. Mebendazole inhibits tumor growth and prevents lung metastasis in models of advanced thyroid cancer. Endocr. Relat. Cancer 2020, 27, 123–136.
- Mansoori, S.; Fryknäs, M.; Alvfors, C.; Loskog, A.; Larsson, R.; Nygren, P. A phase 2a clinical study on the safety and efficacy of individualized dosed Mebendazole in patients with advanced gastrointestinal cancer. Sci. Rep. 2021, 26, 8981.
- Choi, H.S.; Ko, Y.S.; Jin, H.; Kang, K.M.; Ha, I.B.; Jeong, H.; Song, H.N.; Kim, H.J.; Jeong, B.K. Anticancer Effect of Benzimidazole Derivatives, Especially Mebendazole, on Triple-Negative Breast Cancer (TNBC) and Radiotherapy-Resistant TNBC In Vivo and In Vitro. Molecules 2021, 24, 5118.
- Rushworth, L.K.; Hewit, K.; Munnings-Tomes, S.; Somani, S.; James, D.; Shanks, E.; Dufès, C.; Straube, A.; Patel, R.; Leung, H.Y. Repurposing screen identifies Mebendazole as a clinical candidate to synergise with docetaxel for prostate cancer treatment. Br. J. Cancer 2020, 122, 517–527.
- Williamson, T.; de Abreu, M.C.; Trembath, D.G.; Brayton, C.; Kang, B.; Mendes, T.B.; de Assumpção, P.P.; Cerutti, J.M.; Riggins, G.J. Mebendazole disrupts stromal desmoplasia and tumorigenesis in two models of pancreatic cancer. Oncotarget 2021, 6, 1326–1338.
- Elayapillai, S.; Ramraj, S.; Benbrook, D.M.; Bieniasz, M.; Wang, L.; Pathuri, G.; Isingizwe, Z.R.; Kennedy, A.L.; Zhao, Y.D.; Lightfoot, S.; et al. Potential and mechanism of Mebendazole for treatment and maintenance of ovarian cancer. Gynecol. Oncol. 2021, 160, 302–311.
- Hegazy, S.K.; El-Azab, G.A.; Zakaria, F.; Mostafa, M.F.; El-Ghoneimy, R.A. Mebendazole; from an antiparasitic drug to a promising candidate for drug repurposing in colorectal cancer. Life Sci. 2022, 15, 120536.
- Petersen, J.S.S.M.; Baird, S.K. Treatment of breast and colon cancer cell lines with anti-helmintic benzimidazoles mebendazole or albendazole results in selective apoptotic cell death. J. Cancer Res. Clin. Oncol. 2021, 147, 2945–2953.
- Simbulan-Rosenthal, C.M.; Dakshanamurthy, S.; Gaur, A.; Chen, Y.S.; Fang, H.B.; Abdussamad, M.; Zhou, H.; Zapas, J.; Calvert, V.; Petricoin, E.F.; et al. The repurposed anthelmintic Mebendazole in combination with trametinib suppresses refractory NRASQ61K melanoma. Oncotarget 2017, 21, 12576–12595.
- Zhang, F.; Li, Y.; Zhang, H.; Huang, E.; Gao, L.; Luo, W.; Wei, Q.; Fan, J.; Song, D.; Liao, J.; et al. Anthelmintic mebendazole enhances cisplatin’s effect on suppressing cell proliferation and promotes differentiation of head and neck squamous cell carcinoma (HNSCC). Oncotarget 2017, 21, 12968–12982.
- Freisleben, F.; Modemann, F.; Muschhammer, J.; Stamm, H.; Brauneck, F.; Krispien, A.; Bokemeyer, C.; Kirschner, K.N.; Wellbrock, J.; Fiedler, W. Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 10670.
- Sawanyawisuth, K.; Williamson, T.; Wongkham, S.; Riggins, G.J. Effect of the antiparasitic drug Menbendaziole on cholangiocarcinoma growth. Southeast Asian J. Trop. Med. Public Health 2014, 45, 1264–1270.
- Bai, R.Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic Mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-Oncol. 2011, 13, 974–982.
- Ren, L.W.; Li, W.; Zheng, X.J.; Liu, J.Y.; Yang, Y.H.; Li, S.; Zhang, S.; Fu, W.Q.; Xiao, B.; Wang, J.H.; et al. Author Correction: Benzimidazoles induce concurrent apoptosis and pyroptosis of human glioblastoma cells via arresting cell cycle. Acta Pharmacol. Sin. 2022, 15, 194–208.
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14.
- De Witt, M.; Gamble, A.; Hanson, D.; Markowitz, D.; Powell, C.; Al Dimassi, S.; Atlas, M.; Boockvar, J.; Ruggieri, R.; Symons, M. Repurposing mebendazole as a replacement for vincristine for the treatment of brain tumors. Mol. Med. 2017, 23, 50–56.
- Dakshanamurthy, S.; Issa, N.T.; Assefnia, S.; Seshasayee, A.; Peters, O.J.; Madhavan, S.; Uren, A.; Brown, M.L.; Byers, S.W. Predicting new indications for approved drugs using a proteochemometric method. J. Med. Chem. 2012, 55, 6832–6848.
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478.
- Bai, R.Y.; Staedtke, V.; Rudin, C.M.; Bunz, F.; Riggins, G.J. Effective treatment of diverse medulloblastoma models with Mebendazole and its impact on tumor angiogenesis. Neuro-Oncol. 2015, 17, 545–554.
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 20, 8–20.
- Larsen, A.R.; Bai, R.-Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the antihelmintic Mebendazole as a hedgehog inhibitor. Mol. Cancer Ther. 2015, 14, 3–13.
- Bodhinayake, I.; Symons, M.; Boockvar, J.A. Repurposing mebendazole for the treatment of medulloblastoma. Neurosurgery 2015, 76, N15–N16.
- Skibinski, C.G.; Williamson, T.; Riggins, G.J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neurooncol. 2018, 140, 529–538.
- Markowitz, D.; Ha, G.; Ruggieri, R.; Symons, M. Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism. Onco Targets Ther. 2017, 24, 5633–5642.