1. Physiopathology of Glioma Development
Diffuse gliomas represent 80% of malignant brain tumors [
25]. The standard classification of brain tumors by the World Health Organization (WHO, Geneva, Switzerland) is based on their mitotic activity, proliferation, and degree of necrosis; they are identified in four grades:
-
Grade I gliomas are low-grade benign lesions, such as pilocytic astrocytomas, that have limited proliferation and are frequent in children; however, they might acquire higher levels of malignancy. Surgical resection is a good therapeutic option in most cases.
-
Grade II gliomas have low-grade but infiltrative lesions that tend to show higher recurrence after surgical resection.
-
Grade III gliomas have intermediate- to high-grade lesions with atypia, higher mitotic activity, and evidence of malignancy. Patients with these tumors usually are prescribed additional radiation as well as chemotherapy.
-
Grade IV tumors are high-grade malignant lesions with higher mitotic activity, microvascular proliferation, high necrosis, and the worst prognosis.
In 2016, CNS WHO released the latest update of tumor classification on the basis of the latest studies on molecular characterization [
26]. Genomic sequencing efforts provided unprecedented insight into the genomic aberrations and cellular signaling mechanisms that drive these cancers, and discoveries from these efforts translated into novel diagnostic algorithms, biomarkers, and therapeutic strategies in neuro-oncology. These sequencing efforts helped revise WHO classification of nervous system tumors, and histologic and molecular parameters were incorporated into the diagnostic catalog [
26,
27]. Several diagnoses were restructured (diffuse gliomas, medulloblastomas, and embryonal tumors), and novel tumor entities were introduced. For this purpose, researchers at WHO used the datasets generated by The Cancer Genome Atlas (TCGA) and related gene expression and DNA methylation signatures with prognosis. Compared to the previous release, the principal change in the new classification is the relevant reorganization of diffuse gliomas, according to the IDH status, co-deletion of chromosome arms 1p and 19q, and MGMT methylation. The status of isocitrate dehydrogenase 1 and 2 (IDH1/2) enzymes are key reference genes for brain tumor classification. Mutations of IDH1 and IDH2 lead to a favorable outcome [
28] and have more prolonged overall survival in comparison to those with IDH wild type. High-grade gliomas with IDH wild type have shorter overall survival, while low-grade gliomas display a poor glioblastoma (GBM)-like trajectory. The new diagnostic criterion for oligodendroglioma is the allelic loss of chromosomes 1p and 19q [
29]. Although MGMT is not considered a valid predictive biomarker in diffuse gliomas, it is a prognostic biomarker (
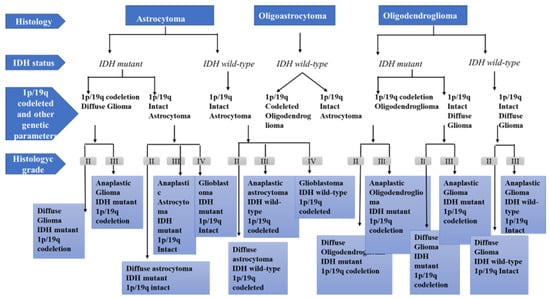
Figure 1) [
30].
Figure 1. Diagnostic algorithm for diffuse gliomas, based on the WHO classification.
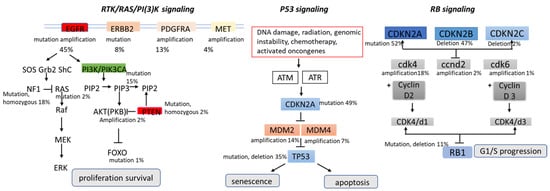
As for signaling pathways, brain tumors manifest aberrations in key cellular pathways that mediate cell growth and replication proliferation, survival, migration, and apoptosis, as observed in other cancers. These pathways are not linear sequences of enzymes but rather complex signaling pathways influenced by multiple growth factors. The Cancer Genome Atlas classified three main signaling pathways in gliomas’ pathogenesis: RTK/RAS/PI3K (receptor tyrosine kinase, RAS, phosphatidylinositol 3-kinase), p53, and retinoblastoma (RB); later, angiogenesis was added as an additional signaling pathway [
26,
31,
32].
The PI3K/AKT/mTOR signal transduction pathway and the Ras/MAPK pathway are hyperactivated in several cancer types, including glioblastoma, and play a key role in tumorigenesis by activating the tumor promoters and inhibiting the tumor suppressors. Several growth factors activate PI3K (e.g., human EGFR family and platelet-derived growth factor receptor (PDGFR) family growth factors). RTKs are frequently activated in malignant gliomas, such as EGFR gene amplification, which occurs in approximately 40% of patients with GBM, and PDGFRA gene amplification, which occurs in up to 16% of GBM [
33,
34,
35,
36]. At the same time, the EGFR variant (EGFRvIII) shows ligand-independent constitutive activation of the receptor. This deletion mutant is observed in approximately 30% to 50% of EGFR-amplified gliomas [
37]. Downstream signaling from growth factor receptors can be activated by loss or mutation in the neurofibromatosis 1 gene (NF1), mutations in KRAS, mutations in PIK3CA (the gene for PI3K), and deletion or loss of heterozygosity of PTEN. PI3K participates in the phosphorylation of AKT and promotes cellular proliferation by inactivating cell cycle inhibitors and promoting cell cycle proteins. The PTEN tumor suppressor gene is inactivated in 40–50% exclusively in primary glioblastomas and usually inhibits the PI3K/Akt pathway [
38].
The Tumor Protein p53 (TP53) tumor suppressor gene encodes a protein that regulates several cellular processes, including cell cycle arrest, the response of cells to DNA damage, senescence, apoptosis, and cell differentiation and neovascularization. The p53 cascade regulates over 2500 genes, 67 of which are involved in tumorigenesis [
39,
40,
41,
42]. After DNA damage, TP53 is activated and induces transcription factors to regulate the expression of downstream effector genes to determine cell fate. The p53 pathway is the most altered in sporadic glioma, with some aberration of p53 signaling found in 87% of tumors [
38]. These aberrations are most prevalent in low-grade glioma of astrocytic lineage and secondary glioblastoma, often concurrent with IDH mutation [
43]. Generally, P53 inactivation in glioblastoma occurs through amplification of MDM2 (mouse double-minute 2) (11%) or MDM4 (4%), deletion of ARF (55%), and mutations of p53 itself [
4]. MDM2 is a primary negative regulator of the p53 pathway; it provides transcriptional inhibition by directly binding p53 and degrades p53 through its ligase activity [
44].
RB is a tumor suppressor gene encoding the retinoblastoma susceptibility protein 1 (RB1). The Rb pathway is an important cell cycle regulator that inhibits the entry of cells through G1 into the S-phase of the cell cycle. The Rb pathway is implicated in the malignant progression of astrocytoma and is commonly altered in glioblastoma (79%). When phosphorylated by cyclin D, cyclin-dependent kinase 4 (CDK4), and CDK6, RB1 will be inactive, resulting in an unregulated progression through the cell cycle and tumor growth [
45] (
Figure 2).
Figure 2. Major signaling pathways involved in the pathogenesis of glioblastomas.
2. Mebendazole: Chemical Structure and Pharmacokinetic Properties
Mebendazole (methyl 5-benzoyl-1H-benzimidazol-2-yl-carbamate) appeared in 1968 as an agent active against a wide range of broad-spectrum anthelmintics applied first to human subjects in 1971 [
46].
The World Health Organization (WHO)-recommended dose of MBZ is 40–50 mg/kg/day for at least 3–6 months and two years for cystic and alveolar echinococcosis, respectively [
60]. The dosage varies according to the type of helminthic infection to treat. Pinworms are treated with a single 100 mg treatment, whereas roundworms or hookworms are treated with 100 mg twice daily for three days. In the case of long-term administration for treating human cystic and alveolar echinococcosis (also known as hydatid disease), the treatment is generally well tolerated. Still, the specific treatment for some patients must be discontinued. For example, in one open-labeled observational study, the patients treated with MBZ for alveolar echinococcosis (average: 24 months) experienced few adverse reactions, and in only three patients (of 17), the treatment was changed to albendazole due to intolerable side effects (reversible alopecia, psychological disturbance, and drop in performance) [
61]. Side effects associated with low- to high-dose regimens include abdominal discomfort, flatulence, diarrhea, neutropenia, marrow aplasia, alopecia, allergic reaction, and elevations in transaminase levels [
60]. However, drug withdrawal spontaneously reverses these relatively rare and mild adverse effects [
23,
62]. One of the most significant adverse effects is hepatotoxicity, although its incidence is infrequent; Tolomei et al. [
63] have reported a case of severe hepatitis after 18 days of MBZ administration in a patient with Gilbert’s syndrome affected by pinworm infestation. After a standard dose of MDZ (2 × 100 mg/d for three days every week) and after short-term treatment, acute hepatitis manifested, with a serum level of ALT (alanine aminotransferase) higher than that observed for higher doses. Hepatic enzymes returned to normal over the following five weeks. Moreover, a case series report supports its safety in breastfeeding, where no evidence of MBZ-related toxicity in infants of lactating mothers has been observed [
64]. MBZ is a teratogen in experimental rats if given in very high doses, although not in rabbits [
31]. In human patients, MBZ is contraindicated during pregnancy [
65]. However, long-term MBZ treatment (50 mg/kg daily for 9–18 months) was safe for children with hydatid disease [
66]. The combination of MBZ (>500 mg) and metronidazole (>500 mg) is prohibited because severe and rare fatal adverse events such as Stevens–Johnson syndrome (or toxic epidermal necrosis) may occur. The risk increased with increasing doses of metronidazole but not MBZ, and there may be a synergistic interaction between MBZ and metronidazole [
67].
Another well-known property of MBZ is its relative insolubility in water and most organic solvents. The absorption rate of oral MBZ in the human intestine is about 1–5% [
51]. The low water solubility can cause a reduction in drug absorption that, combined with the rapid drug degradation, could result in an inadequate in vivo drug concentration and failure to achieve the therapeutic dose. Moreover, oscillation in the drug plasma levels results in unforeseeable levels of bioavailability. All these effects cause a reduction in MBZ’s potency in cancer therapy; thus, modifications enhancing its availability will determine its potential use in oncology. Several researchers suggested strategies to improve the poor aqueous MBZ and anticancer effects [
59].
Alavi and Shahmabadi provide an overview of the strategies adopted to improve low bioavailability. To overcome these limitations, the authors proposed using nanocarrier-based formulations, pro-drug formulations, and solid dispersions (SDs) to improve drug solubility [
68]. To overcome poor aqueous solubility, de la Torre-Iglesias et al. [
69] have prepared re-dispersible microparticles (RDM) containing MBZ for improved oral bioavailability and therapeutic activity using low doses of MBZ (5 mg/kg). These formulations could optimize anthelmintic efficacy and enhance MBZ concentrations in muscle and cysts. This aspect is crucial for inoperable or disseminated cases of hydatidosis or neuro-cysticercosis.
Furthermore, low doses of MBZ lead to low toxicity in humans. Münst et al. [
70] have demonstrated that bioavailability may significantly improve by coadministration with a high-fat meal. Plasma concentrations remained at 17 nmol/L in three fasting volunteers treated with 1.5 g doses. Instead, when the treated volunteers had a standard breakfast, the plasma concentration rose to 91,112 and 142 nmol/L within 2 to 4 h. In another clinical study, Braithwaite et al. [
55] showed that MBZ plasma concentration-time profiles differed considerably between patients after oral administration of MBZ. The authors monitored the plasma concentrations of MBZ and its metabolites in twelve patients after receiving 10 mg/kg, in subjects receiving their first dose of MBZ, and in subjects with chronic treatment. The elimination half-lives ranged from 2.8–9.0 h; the peak plasma concentrations ranged from 17.7 to 116.2 ng/mL for subjects receiving their initial dose and from 99.4 to 500.2 ng/mg for subjects on chronic treatment. Studies reported by Alavi et al. [
68] showed that the plasma AUCTs for the main metabolites of MBZ (methyl 5-(α-hydroxybenzyl)-2-benzimidazole carbamate and 2-amino-5 benzoylbenzimidazole) were about five times larger than the plasma MBZ AUCT found in patients on chronic therapy, confirming that the gastrointestinal tract poorly absorbs MBZ.
In patients on chronic MBZ therapy, MBZ concentrations found in both liver tissue and parasite tissue were significantly higher than in adipose tissue. While the MBZ concentration in liver was higher, that in other tissues was lower than the plasma Cmax detected for each patient. To reduce the wide inter- and intra-individual variation in systemic availability of MBZ, the drug should be taken with food. For intravenous administration, no guidelines can be provided because there are no data on the pharmacokinetics of MBZ after intravenous infusion. Bekhti et al. [
57] and Luder et al. [
58] proved that MBZ produces pharmacodynamic effects possibly exaggerated by pharmacokinetic interactions with other agents. In that paper, they showed that cimetidine increases plasma levels of the MBZ (maximum serum levels rose from 55.7 ± 30.2 ng/mL [0.19 ± 0.10 μM] to 82.3 ± 41.8 ng/mL [0.28 ± 0.14 μM], on 1.5 g of MBZ following chronic dosing of cimetidine at 400 mg three times a day for 30 days). The available evidence suggests that the increase in Cmax of MBZ, caused by concurrent use of cimetidine, would influence the therapeutic actions of MBZ, probably due to inhibition of hepatic first-pass cytochrome P450-mediated metabolism of the MBZ. However, what remains unclear is whether such an increase in Cmax would predispose a patient to adverse effects. Due to such an interaction, clinicians should closely monitor patients receiving this combination for signs of adverse effects of MBZ and choose alternative agents when possible. In addition, Corti et al. [
56] investigated the effect of ritonavir (protease inhibitor of the CYP3A system) on the pharmacokinetics of MBZ under single-dose and steady-state conditions. They observed that the pharmacokinetic parameters of MBZ did not change in the short-term administration of ritonavir. In contrast, long-term administration of ritonavir resulted in significant changes in MBZ disposition, with a substantial decrease in AUC and Cmax. In addition, three polymorphs, A, B, and C, of MBZ exist, showing different solubility, toxicity, and therapeutic effects in anthelmintic applications [
71,
72,
73]. Ren-Yuan Bai et al. [
74] determined the pharmacokinetics of MBZ-A, B, and C and their concentrations in the brain and brain tumor distribution after oral MBZ administration on GL261 tumor-bearing mice. They suggest that MBZ-C is the most efficacious polymorph in brain tumor therapy with limited toxicity. Moreover, they demonstrated that the combination of Mebendazole-C with elacridar (P-glycoprotein inhibitor) could significantly improve the efficacy for future Phase I clinical trials of high-grade glioma and medulloblastoma. The high intra- and inter-patient variability may be essential to evaluate the response of MBZ as a potential anticancer therapy. Therefore, further research should elucidate the pharmacokinetic and dynamic profiles of upcoming formulations. To this end, monitoring the levels of MBZ and its metabolites should become standard practice in clinical treatments. Currently, preclinical evidence shows that chronic and high-dose schedules achieve plasma levels in the range required for clinical activity.
3. Preclinical Evidence of Mebendazole in Brain Cancer—In Vitro and In Vivo
Anthelmintic drugs have gained attention in the last two decades as potential anticancer agents due to their interactivity with microtubules [
23,
24,
75,
76]. In particular, a wide range of cancer cells and animal models showed the possible effect of MBZ in inhibiting tumor cell growth through its ability to inhibit tubulin polymerization, leading to a lethal effect in rapidly dividing cells. The potential effect of MBZ in inhibiting cancer cell growth has been described in thyroid [
77], gastrointestinal [
78], breast [
79], prostate [
80], pancreatic [
81], ovarian [
82], colorectal [
83,
84], melanoma [
85], head and neck [
86], leukemia [
87], and bile duct cancer [
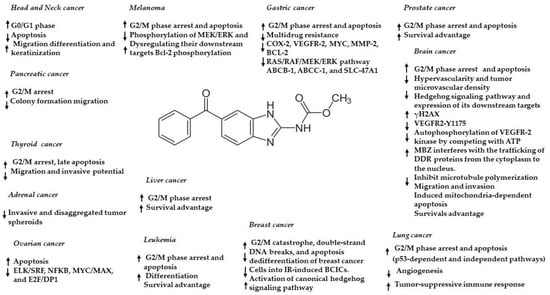
88]. Moreover, MBZ can modulate various cancer-associated pathways, including MAPK14, MEK-ERK, C-MYC, USP5/c-Maf, TNIK, XIAP, ELK/SRF, NFKB, MYC/MAX, E2F/DP1, TGF/SMAD, AP1, and STAT1/2, dependent on the specific cancer model (
Figure 3) [
23,
24]. In addition, MBZ is relatively non-toxic for normal cells, whereas it increases its specific sensitivity in cancer cells. Regarding brain cancer, several studies demonstrated the antitumor properties of MBZ. In 2011, routine animal studies unexpectedly showed that fenbendazole (a benzimidazole anthelmintic used to treat pinworm infection) inhibited brain tumor engraftment. Based on this hint, Ren-Yuan Bai et al. [
89] proved that MBZ offers an auspicious opportunity for the clinical application of GBM; their research with in vitro and in vivo experiments with benzimidazoles identified for the first time MBZ as the most promising drug for GBM therapy. In detail, they observed that MBZ induced apoptosis in GBM cell lines. The IC
50 was 0.24 μM in the GL261 mouse glioma line and 0.1 μM in the 060919 human GBM. In vitro activity was also correlated with significant inhibition of tubulin polymerization at 0.1 µM. They also proved that MBZ significantly extended mean survival to 65 days, compared with 48 days for the control group in syngeneic and xenograft orthotopic mouse glioma models. Moreover, they showed that the combination of MBZ plus temozolomide (TMZ) extends survival further than TMZ alone in the GL261 mouse model. Other diverse mechanisms of MBZ in CNS tumors have been proposed. In particular, Ren LW et al. [
90] have suggested that all benzimidazole compounds (flubendazole, Mebendazole, fenbendazole) could inhibit the proliferation and metastasis of GBM cells regulating cell migration, cell cycle, programmed cell death, and other biological processes. Furthermore, MBZ inhibited the migration and invasion of GBM cells and regulated the expression of crucial (epithelial-to-mesenchymal transition) EMT markers, showing that MBZ could inhibit the metastasis of GBM. It also dose-dependently arrested the cell cycle at the G2/M phase of GBM cells through the P53/P21/cyclin B1 pathway.
Figure 3. Anticancer effects and mechanisms of action of MBZ in different cancers. ↑ upregulation/activation; ↓ downregulation/inhibition.
The greatest challenge facing any CNS-targeted drug discovery program is the effective penetration of the blood–brain barrier (BBB). The limited ability of cancer therapeutics to accumulate in the tumor is the major obstacle to improving brain cancer therapy [
91]. It is estimated that only ~2% of small-molecule drugs can effectively cross the BBB. MBZ’s small size (295 Daltons) and lipophilic property favor brain penetration. Depending on crystallization conditions, MBZ can form three different polymorphs, A, B, and C, which display significant differences in bioavailability. Ren-Yuan Bai et al. [
74] studied MBZ’s brain penetration and pharmacokinetics and the therapeutic differences of the three polymorphic forms (A, B, C) on intracranial murine GL261 glioma allografts and human medulloblastoma D425 xenografts. The authors determined that MBZ-A showed low blood and brain concentrations and no antitumoral efficacy; however, MBZ-B and MBZ-C are therapeutically favorable for brain tumor therapy. Polymorph C demonstrated the highest penetration capability in the brain tissue and tumor. Furthermore, combination therapy of elacridar (P-glycoprotein (P-gp) inhibitor) and MBZ-C increased the survival in GL261 syngeneic glioma and D425 xenograft medulloblastoma models. In addition, De Witt M et al. [
92] examined the mechanisms of tumor cell-killing of MBZ and cell viability compared with those of vincristine on GL261 glioblastoma cells. They demonstrated that MBZ and vincristine have similar inhibitory effects on cell viability and microtubule polymerization. They also compared the therapeutic efficacies and toxicities of MBZ and vincristine in the GL261-C57BL/6 syngeneic orthotopic mouse model, showing that MBZ is more effective than vincristine and provides significant survival.
Recently, Dakshanamurthy and colleagues [
93] applied a computational proteo-chemometric method to a library of FDA-approved compounds and identified MBZ as a potential inhibitor of vascular endothelial growth factor receptor 2 (VEGFR2), which suggested a possible role of MBZ in interfering with tumor angiogenesis. VEGF is an essential pathway in angiogenesis-related glioma pathology; in glioblastoma, VEGF is upregulated and therefore induces angiogenesis with consequent production of dysfunctional and immature vessels, associated with significant edema and destruction of the BBB [
94]. For this reason, therapeutic inhibition of VEGFR-2 action has a significant impact on restricting cancer growth. Numerous studies confirm that MBZ can suppress the autophosphorylation of VEGFR-2 kinase by competing with ATP. In this context, Ren-Yuan Bai et al. [
95] tested the drug on a panel of eight medulloblastoma cell lines, obtaining IC
50 for cell growth of 0.13–1 µM. They found that MBZ also inhibited vascular endothelial growth factor receptor 2 (VEGFR2) autophosphorylation at 1–10µM, while it cultured HUVECs with an IC
50 of 4.3 µM. They demonstrated that MBZ selectively exhibited tumor angiogenesis but not normal brain vasculatures in orthotopic medulloblastoma models and interfered with VEGFR2 activity in multiple models. MBZ was able to prolong the median survival from 21 days to 48 days in the D425 xenograft medulloblastoma. Analysis of tumor sections from treated mice revealed a significant reduction of tumor angiogenesis, while the microvessel density in the normal brain parenchyma was unaffected.
A growing body of evidence suggests that activated Hedgehog (Hh) signaling is preclinically responsive to MBZ. Some studies indicate that the aberrant activation of Hh signaling leads to tumor cell growth, proliferation, and invasion. Therefore, the development of therapy targeting Hh signaling is an attractive and validated therapeutic strategy for treating a wide range of cancers [
96]. Larsen et al. [
97] found that MBZ is a potent inhibitor of the Hh signaling cascade in human medulloblastoma cultured cell lines and decreased GLI1 expression with an IC
50 of 516 nM. Cell proliferation was inhibited at a concentration as low as 100 nM, and viability was significantly impaired at one µM. In the same paper, the authors suggested that MBZ treatment prevented the primary cilium formation. This microtubule-based organelle functions as a signaling hub for Hh pathway activation, decreasing the expression of downstream Hh pathway effectors and the proliferation and survival of human medulloblastoma cells with constitutive Hh activation. The activity of MBZ in vivo was then assessed in a DAOY intracranial mouse xenograft. Treatment significantly increased survival from 75 days in the control group to 94 days in the group administered MBZ 25 mg/kg and 113 days in the 50 mg/kg group. Along this same line, the study by Bodhinayake et al. [
98] highlighted that using orthotopic models of medulloblastoma (a genetic model of the sonic hedgehog (SHH)allograft, SHH vismodegib-resistant, and D425 implanted into cerebellum) the MBZ treatment induced markedly extended survival of 150%, 100%, and 129%, respectively.
MBZ can also sensitize cancer cells to conventional therapy, such as chemotherapeutics and radiation, enhancing their combined antitumor potential, confirming that MBZ may be useful as an adjuvant therapeutic combined with traditional chemotherapy. Skibinski et al. [
99] demonstrated that MBZ administration alone or combined with radiation effectively extended survival in preclinical models of malignant meningioma for the first time. The human meningioma cell lines treated with MBZ alone had IC
50 values in the 0.26–0.42 μM range, similar to those observed in medulloblastoma and glioblastoma. The authors also described a synergistic effect when combining MBZ therapy with radiation in an intracranial mouse model of malignant meningioma. This combination increased median survival and delayed tumor growth by inducing apoptosis via the caspase-3/7 pathway, decreasing cell proliferation and reducing levels of the angiogenesis marker CD31. Another study observed significant improvement in the radiosensitization of glioma cells (GL261 and GBM14) after MBZ treatment. Markowitz et al. [
100] treated GL261 and GBM14 glioma cells with MBZ 3–9 h post-IR; this experiment demonstrated that MBZ could sensitize cancer cells to IR independently of the induction of mitotic arrest.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24021334