Therapeutic proteins, including monoclonal antibodies, single chain variable fragment (ScFv), crystallizable fragment (Fc), and fragment antigen binding (Fab), have accounted for one-third of all drugs on the world market. In particular, these medicines have been widely used in ocular therapies in the treatment of various diseases, such as age-related macular degeneration, corneal neovascularization, diabetic retinopathy, and retinal vein occlusion. The formulation of these biomacromolecules is challenging due to their high molecular weight, complex structure, instability, short half-life, enzymatic degradation, and immunogenicity, which leads to the failure of therapies. Various efforts have been made to overcome the ocular barriers, providing effective delivery of therapeutic proteins, such as altering the protein structure or including it in new delivery systems. These strategies are not only cost-effective and beneficial to patients but have also been shown to allow for fewer drug side effects. Researchers discuss several factors that affect the design of formulations and the delivery of therapeutic proteins to ocular tissues, such as the use of injectable micro/nanocarriers, hydrogels, implants, iontophoresis, cell-based therapy, and combination techniques. In addition, other approaches are briefly discussed, related to the structural modification of these proteins, improving their bioavailability in the posterior segments of the eye without affecting their stability. Future research should be conducted toward the development of more effective, stable, noninvasive, and cost-effective formulations for the ocular delivery of therapeutic proteins. In addition, more insights into preclinical to clinical translation are needed.
- ocular diseases
- sustained ocular delivery
- therapeutic proteins
1. Introduction
2. Routes of Ocular Drug Administration
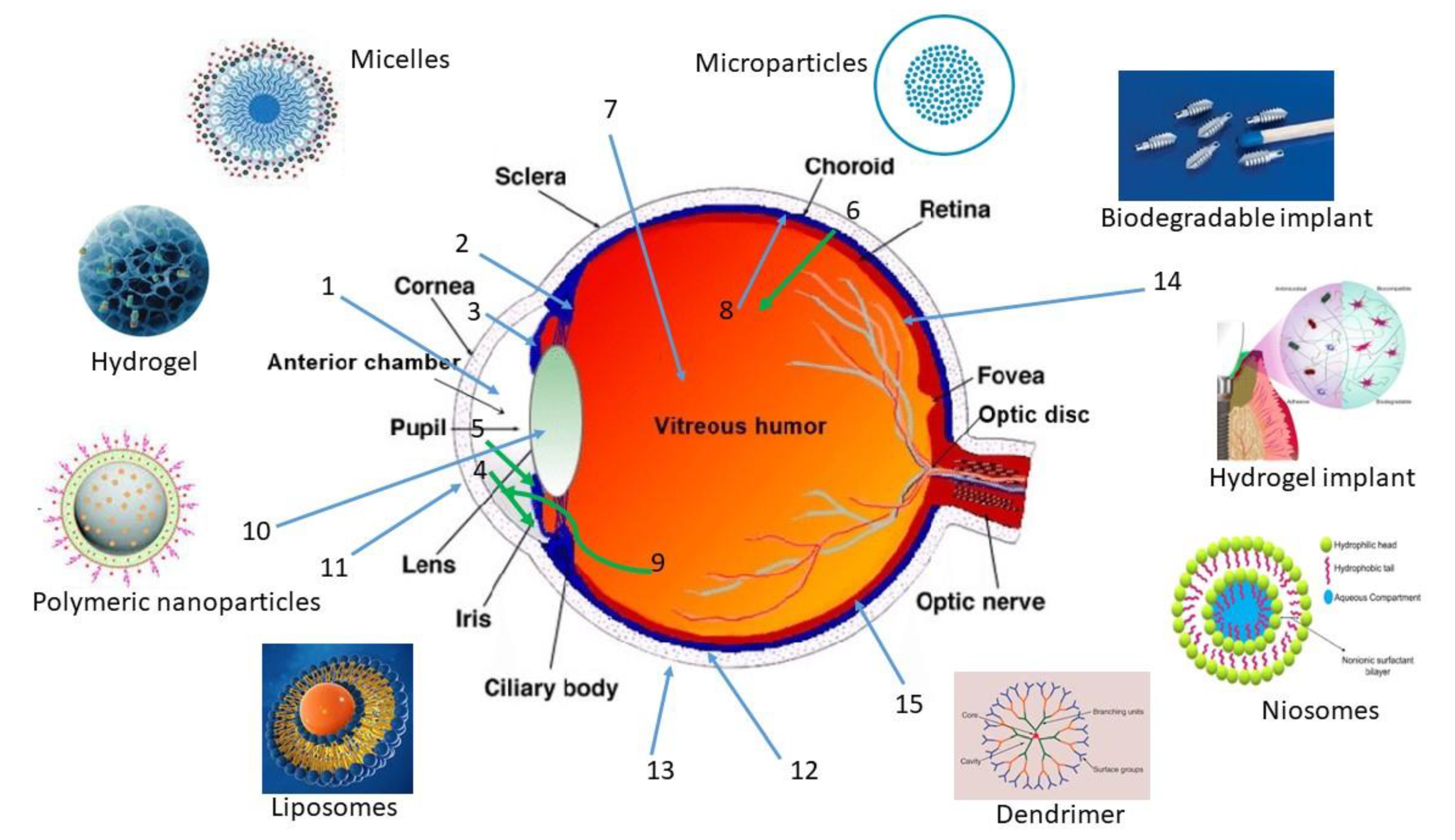
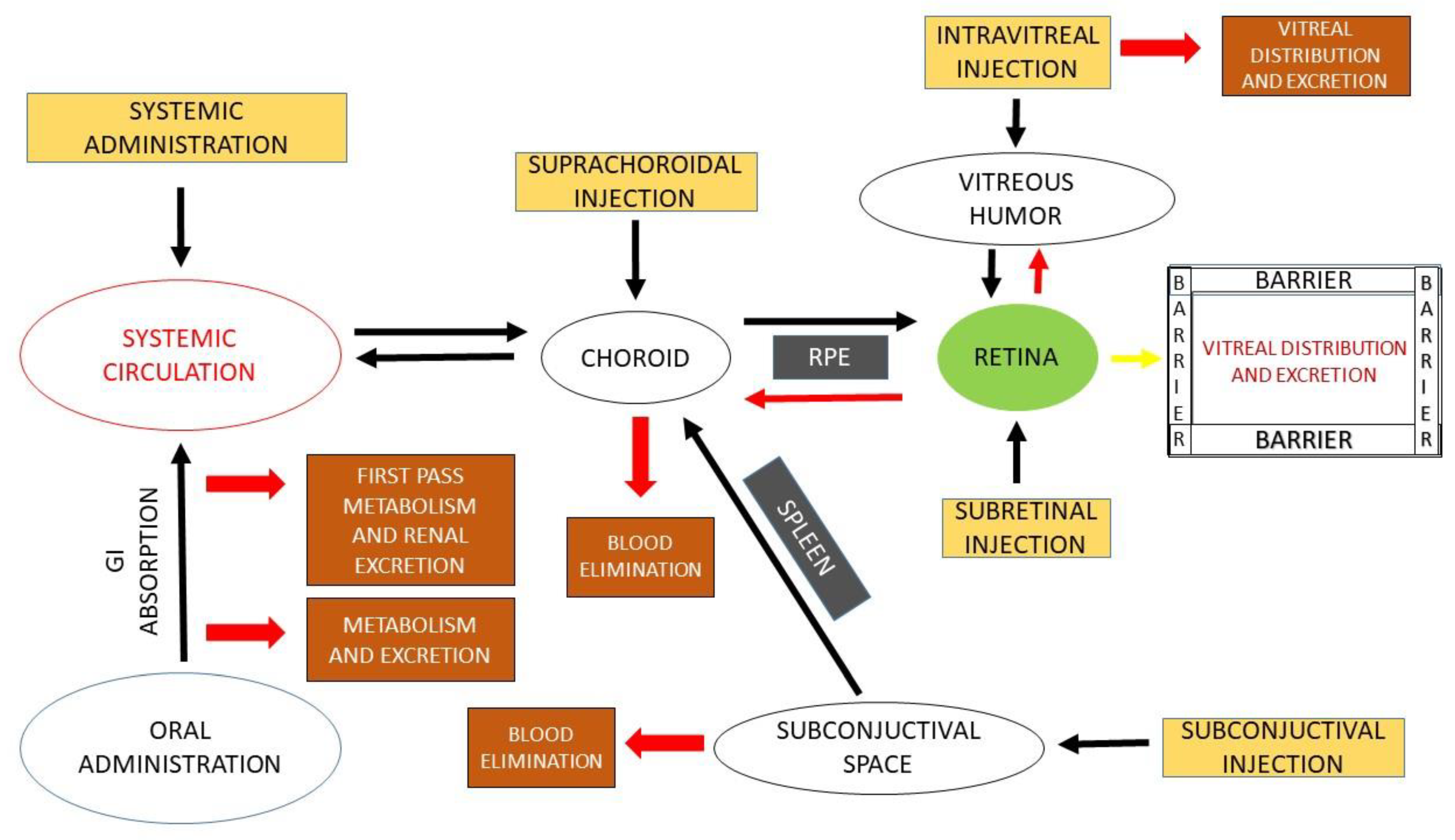
2.1. Intraocular
- (a)
-
Intravitreal
- (b)
-
Subretinal
- (c)
-
Suprachoroidal
2.2. Periocular
3. Ocular Barriers and Approaches to Ocular Administration
3.1. Ocular Barriers
3.1.1. Tissue Conditions
3.1.2. Physicochemical Characteristics of Drug Molecules
3.1.3. Viscosity and pH of the Formulation
3.1.4. Protein Binding
3.1.5. Enzymatic Degradation
3.2. Use of Penetration Enhancers
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15010205
References
- Sharma, D.S.; Wadhwa, S.; Gulati, M.; Ramanunny, A.K.; Awasthi, A.; Singh, S.K.; Khursheed, R.; Corrie, L.; Chitranshi, N.; Gupta, V.K.; et al. Recent advances in intraocular and novel drug delivery systems for the treatment of diabetic retinopathy. Expert Opin. Drug Del. 2021, 18, 553–576.
- Kim, H.M.; Woo, S.J. Ocular Drug Delivery to the Retina: Current Innovations and Future Perspectives. Pharmaceutics 2021, 13, 108.
- Bourne, R.R.A.; Flaxman, S.R.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; Leasher, J.; Limburg, H.; et al. Vision Loss Expert Group. Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e888–e897.
- Claudio, F.; Francesco, B.; Michele, R.; Giovanni, A. Intravitreal Therapy for Diabetic Macular Edema: An Update. J. Ophthalmol. 2021, 2021, 6654168.
- Aiello, L.P. The potential role of PKC b in diabetic retinopathy and macular edema. Survey Ophthalmol. 2002, 47 (Suppl. S2), 263–269.
- Alqahtani, F.Y.; Aleanizy, F.S.; El Tahir, E.; Alquadeib, B.T.; Alsarra, I.A.; Alanazi, J.S.; Abdelhady, H.G. Preparation, characterization, and antibacterial activity of diclofenac-loaded chitosan nanoparticles. Saudi Pharm. J. 2019, 27, 82–87.
- Hu, J.; Zhang, Y.; Li, X.; Han, W.; Zheng, J.; Yang, G.; Xu, A. Combination of Intrastromal and Intracameral Injections of Amphotericin B in the Treatment of Severe Fungal Keratitis. J. Ophthalmol. 2016, 2016, 3436415.
- Patel, P.B.; Shastri, D.H.; Shelat, P.K.; Shukla, A.K. Ophthalmic drug delivery system: Challenges and approaches. Syst. Rev. Pharm. 2010, 1, 113–120.
- Liu, W.; Borrell, M.A.; Venerus, D.C.; Mieler, W.F.; Kang-Mieler, J.J. Characterization of Biodegradable Microsphere-Hydrogel Ocular Drug Delivery System for Controlled and Extended Release of Ranibizumab. Transl. Vis. Sci. Technol. 2019, 8, 12.
- Narayana, S.; Ahmed, M.G.; Gowda, J.B.H.; Shetty, P.K.; Nasrine, A.; Thriveni, M.; Noushida, N.; Sanjana, A. Recent advances in ocular drug delivery systems and targeting VEGF receptors for management of ocular angiogenesis: A comprehensive review. Future J. Pharm. Sci. 2021, 7, 186.
- Chen, W.; Yung, B.C.; Qian, Z.; Chen, X. Improving Long-Term Subcutaneous Drug Delivery by Regulating Material-Bioenvironment Interaction. Adv. Drug Deliv. Rev. 2018, 127, 20–34.
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.S.; Mitra, A.K. Approaches for Enhancing Oral Bioavailability of Peptides and Proteins. Int. J. Pharm. 2013, 447, 75–93.
- Faulds, D.; Goa, K.L.; Benfield, P. Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs 1993, 45, 953–1040.
- Brown, L.R. Commercial Challenges of Protein Drug Delivery. Expert Opin. Drug Deliv. 2005, 2, 29–42.
- Bhattacharya, M.; Sadeghi, A.; Sarkhel, S.; Hagström, M.; Bahrpeyma, S.; Toropainen, E.; Auriola, S.; Urtti, A. Release of functional dexamethasone by intracellular enzymes: A modular peptide-based strategy for ocular drug delivery. J. Control. Release 2020, 327, 584–594.
- Haddadzadegan, S.; Dorkoosh, F.; Bernkop-Schnürch, A. Oral delivery of therapeutic peptides and proteins: Technology landscape of lipid-based nanocarriers. Adv. Drug Deliv. Rev. 2022, 182, 114097.
- Lin, J. Pharmacokinetics of Biotech Drugs: Peptides, Proteins and Monoclonal Antibodies. Curr. Drug Metab. 2009, 10, 661–691.
- Ahmed, I.; Patton, T.F. Disposition of Timolol and Inulin in the Rabbit Eye Following Corneal versus Non-Corneal Absorption. Int. J. Pharm. 1987, 38, 9–21.
- Donovan, M.D.; Flynn, G.L.; Amidon, G.L. Absorption of Polyethylene Glycols 600 Through 2000: The Molecular Weight Dependence of Gastrointestinal and Nasal Absorption. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1990, 7, 863–868.
- Shen, W.; Matsui, T. Intestinal Absorption of Small Peptides: A Review. Int. J. Food Sci. Technol. 2019, 54, 1942–1948.
- Xu, Q.; Boylan, N.J.; Suk, J.S.; Wang, Y.Y.; Nance, E.A.; Yang, J.C.; McDonnell, P.J.; Cone, R.A.; Duh, E.J.; Hanes, J. Nanoparticle Diffusion in, and Microrheology of, the Bovine Vitreous Ex Vivo. J. Control. Release 2013, 167, 76–84.
- Xu, J.; Heys, J.J.; Barocas, V.H.; Randolph, T.W. Permeability and Diffusion in Vitreous Humor: Implications for Drug Delivery. Pharm. Res. 2000, 17, 664–669.
- Balachandran, R.K.; Barocas, V.H. Contribution of Saccadic Motion to Intravitreal Drug Transport: Theoretical Analysis. Pharm. Res. 2011, 28, 1049–1064.
- Käsdorf, B.T.; Arends, F.; Lieleg, O. Diffusion Regulation in the Vitreous Humor. Biophys. J. 2015, 109, 2171–2181.
- Nakano, M.; Lockhart, C.M.; Kelly, E.J.; Rettie, A.E. Ocular Cytochrome P450s and Transporters: Roles in Disease and Endobiotic and Xenobiotic Disposition. Drug Metab. Biophys. J. Rev. 2014, 46, 247–260.
- Urtti, A. Nanostructures Overcoming the Ocular Barrier: Drug Delivery Strategies; Chapter 4.2.; Royal Society of Chemistry: London, UK, 2012; pp. 190–204.
- DiCarlo, J.E.; Mahajan, V.B.; Tsang, S.H. Gene therapy and genome surgery in the retina. J. Clin. Investig. 2018, 128, 2177–2188.
- Sørensen, N.B. Subretinal Surgery: Functional and Histological Consequences of Entry into the Subretinal Space. Acta Ophthalmol. 2019, 97, 1–23.
- Subrizi, A.; del Amo, E.M.; Korzhikov-Vlakh, V.; Tennikova, T.; Ruponen, M.; Urtti, A. Design Principles of Ocular Drug Delivery Systems: Importance of Drug Payload, Release Rate, and Material Properties. Drug Discov. Today. 2019, 24, 1446–1457.
- Bachu, R.D.; Chowdhury, P.; Al-Saedi, Z.H.F.; Karla, P.K.; Boddu, S.H.S. Ocular Drug Delivery Barriers-Role of Nanocarriers in the Treatment of Anterior Segment Ocular Diseases. Pharmaceutics 2018, 10, 28.
- Muheem, A.; Shakeel, F.; Jahangir, M.A.; Anwar, M.; Mallick, N.; Jain, G.K.; Warsi, M.H.; Ahmad, F.J. A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharm. J. 2016, 24, 413–428.
- Leclercq, B.; Mejlachowicz, D.; Behar-Cohen, F. Ocular Barriers and Their Influence on Gene Therapy Products Delivery. Pharmaceutics 2022, 14, 998.
- Tao, Y.; Li, X.X.; Jiang, Y.R.; Bai, X.B.; Wu, B.D.; Dong, J.Q. Diffusion of macromolecule through retina after experimental branch retinal vein occlusion and estimate of intraretinal barrier. Curr. Drug Metab. 2007, 8, 151–156.
- Jackson, T.L.; Antcliff, R.J.; Hillenkamp, J.; Marshall, J. Human retinal molecular weight exclusion limit and estimate of species variation. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2141–2146.
- Blessing, C.I.; Charis, R.; Roel, F.M.; Mio, T.; Mei, C.; Wim, E.H. Hyaluronic Acid-PEG-Based Diels–Alder In Situ Forming Hydrogels for Sustained Intraocular Delivery of Bevacizumab. Biomacromolecules 2022, 23, 1525–7797.
- Burgalassi, S.; Monti, D.; Nicosia, N.; Tampucci, S.; Terreni, E.; Vento, A.; Chetoni, P. Freeze-dried matrices for ocular administration of bevacizumab: A comparison between subconjunctival and intravitreal administration in rabbits. Drug Deliv. Transl. Res. 2018, 8, 461–472.
- Urtti, A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1131–1135.
- Levison, M.E.; Levison, J.H. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect. Dis. Clini. N. Am. 2009, 23, 791–815.
- Del Amo, E.M.; Urtti, A. Rabbit as an animal model for intravitreal pharmacokinetics: Clinical predictability and quality of the published data. Exp. Eye. Res. 2015, 137, 111–124.
- Sidman, R.L.; Li, J.; Lawrence, M.; Hu, W.; Musso, G.F.; Giordano, R.J.; Cardo-Vila, M.; Pasqualini, R.; Arap, W. The peptidomimetic Vasotide targets two retinal VEGF receptors and reduces pathological angiogenesis in murine and nonhuman primate models of retinal disease. Sci. Transl. Med. 2015, 7, 309.
- Pescina, S.; Ferrari, G.; Govoni, P.; Macaluso, C.; Padula, C.; Santi, P.; Nicoli, S. In-vitro permeation of bevacizumab through human sclera: Effect of iontophoresis application. J. Pharm. Pharmacol. 2010, 62, 1189–1194.
- Swami, R.; Shahiwala, A. Impact of physiochemical properties on pharmacokinetics of protein therapeutics. Eur. J. Drug Metab. Pharmacokinetics. 2013, 38, 231–239.
- Kuo, T.T.; Baker, K.; Yoshida, M.; Qiao, S.W.; Aveson, V.G.; Lencer, W.I.; Blumberg, R.S. Neonatal Fc receptor: From immunity to therapeutics. J. Clin. Immunol. 2010, 30, 777–789.
- Gaudana, R.; Ananthula, H.K.; Parenky, A.; Mitra, A.K. Ocular Drug Delivery. AAPS J. 2010, 12, 348–360.
- Edelhauser, H.F.; Rowe-Rendleman, C.L.; Robinson, M.R.; Dawson, D.G.; Chader, G.J.; Grossniklaus, H.E. Ophthalmic drug delivery systems for the treatment of retinal diseases: Basic research to clinical applications. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5403–5420.
- Shen, J.; Durairaj, C.; Lin, T.; Liu, Y.; Burke, J. Ocular pharmacokinetics of intravitreally administered brimonidine and dexamethasone in animal models with and without blood-retinal barrier breakdown. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1056–1066.
- Kim, Y.C.; Chiang, B.; Wu, X.; Prausnitz, M.R. Ocular delivery of macromolecules. J. Con. Rel. 2014, 190, 172–181.
- Joseph, R.R.; Venkatraman, S.S. Drug delivery to the eye: What benefits do nanocarriers offer? Nanomedicine 2017, 12, 683–702.
- Raghava, S.; Hammond, M.; Ub, K. Periocular routes for retinal drug delivery. Expert Opin. Drug Deliv. 2004, 1, 99–114.
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Retinal pigment epithelial cells express a functional receptor for glucagon-like peptide-1 (GLP-1). Mediat. Inflamm. 2013, 2013, 975032.
- Zelikin, A.N.; Ehrhardt, C.; Healy, A.M. Materials and methods for delivery of biological drugs. Nat. Chem. 2016, 8, 997–1007.
- Chang, J.H.; Garg, N.K.; Lunde, E.; Han, K.Y.; Jain, S.; Azar, D.T. Corneal neovascularization: An anti-VEGF therapy review. Surv. Ophthalmol. 2012, 57, 415–429.
- Vinores, S.A. Pegaptanib in the treatment of wet, age-related macular degeneration. Int. J. Nanomed. 2006, 1, 263–268.
- Joseph, M.; Trinh, H.M.; Cholkar, K.; Pal, D.; Mitra, A.K. Recent perspectives on the delivery of biologics to back of the eye. Expert Opin. Drug Deliv. 2016, 14, 631–645.
- Xu, L.; Lu, T.; Tuomi, L.; Jumbe, N.; Lu, J.; Eppler, S.; Kuebler, P.; Damico-Beyer, L.A.; Joshi, A. Pharmacokinetics of ranibizumab in patients with neovascular age-related macular degeneration: A population approach. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1616–1624.
- Vaishya, R.D.; Khurana, V.; Patel, S.; Mitra, A.K. Controlled ocular drug delivery with nanomicelles, Wiley interdisciplinary reviews. Nanomed. Nanobiotechnol. 2014, 6, 422–437.
- Moisseiev, E.; Waisbourd, M.; Ben-Artsi, E.; Levinger, E.; Barak, A.; Daniels, T.; Csaky, K.; Loewenstein, A.; Barequet, I.S. Pharmacokinetics of bevacizumab after topical and intravitreal administration in human eyes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2014, 252, 331–337.
- Sharma, Y.R.; Venkatesh, P.; Gogia, V. Aflibercept—How does it compare with other Anti-VEGF Drugs? Aust. J. Clin. Ophthalmol. 2014, 1, 1–8.
- Neri, P.; Lettieri, M.; Fortuna, C.; Zucchi, M.; Manoni, M.; Celani, S.A. Giovannini, Adalimumab (humira) in ophthalmology: A review of the literature. Middle East Afr. J. Ophthalmol. 2010, 17, 290–296.
- Rodrigues, E.B.; Farah, M.E.; Maia, M.; Penha, F.M.; Regatieri, C.; Melo, G.B.; Pinheiro, M.M.; Zanetti, C.R. Therapeutic monoclonal antibodies in ophthalmology. Prog. Ret. Eye Res. 2009, 28, 117–144.
- Khawli, L.A.; Goswami, S.; Hutchinson, R.; Kwong, Z.W.; Yang, J.; Wang, X.; Yao, Z.; Sreedhara, A.; Cano, T.; Tesar, D. Charge variants in IgG1: Isolation, characterization, In vitro binding properties and pharma-cokinetics in rats. MAbs 2010, 2, 613–624.
- Maurice, D.M.; Watson, P.G. The distribution and movement of serum albumin in the cornea. Exp. Eye Res. 1965, 4, 355–363.
- Kim, J.H.; Green, K.; Martinez, M.; Paton, D. Solute permeability of the corneal endothelium and Descemet’s membrane. Exp. Eye Res. 1971, 12, 231–238.
- Olsen, T.W.; Edelhauser, H.F.; Lim, J.I.; Geroski, D.H. Human scleral permeability. Effects of age, cryotherapy, transscleral diode laser, and surgical thinning. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1893–1903.
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672.
- Duvvuri, S.; Majumdar, S.; Mitra, A.K. Drug delivery to the retina: Challenges and opportunities. Expert Opin. Biol. Ther. 2003, 3, 45–56.
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575.
- Li, S.K.; Liddell, M.R.; Wen, H. Effective electrophoretic mobilities and charges of anti-VEGF proteins determined by capillary zone electrophoresis. J. Pharm. Biomed. Anal. 2011, 55, 603–607.
- Kaja, S.; Hilgenberg, J.D.; Everett, E.; Olitsky, S.E.; Gossage, J.; Koulen, P. Effects of dilution and prolonged storage with preservative in a polyethylene container on Bevacizumab (Avastin) for topical delivery as a nasal spray in anti-hereditary hemorrhagic telangiectasia and related therapies. Hum. Antibodies 2011, 20, 95–101.
- Gregoritza, M.; Messmann, V.; Abstiens, K.; Brandl, F.P.; Goepferich, A.M. Controlled antibody release from degradable thermoresponsive hydrogels cross-linked by Diels-Alder chemistry. Biomacromolecules 2017, 18, 2410–2418.
- Jani, R.; Lang, J.; Rodeheaver, D.; Missel, P.; Roehrs, R.; Chowhan, M. Design and Evaluation of Ophthalmic Pharmaceutical Products, Modern Pharmaceutics, 4th ed.; CRC Press: Boca Raton, FL, USA, 2002.
- Ali, Y.; Lehmussaari, K. Industrial perspective in ocular drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1258–1268.
- Subrizi, A.; Toropainen, E.; Ramsay, E.; Airaksinen, A.J.; Kaarniranta, K.; Urtti, A. Oxidative stress protection by exogenous delivery of rhHsp70 chaperone to the retinal pigment epithelium (RPE), a possible therapeutic strategy against RPE degeneration. Pharm. Res. 2015, 32, 211–221.
- Schymkowitz, J.; Rousseau, F. Protein aggregation: A rescue by chaperones. Nat. Chem. Biol. 2016, 12, 58–59.
- Angi, M.; Kalirai, H.; Coupland, S.E.; Damato, B.E.; Semeraro, F.; Romano, M.R. Proteomic Analyses of the Vitreous Humour. Mediat. Inflamm. 2012, 2012, 148039.
- Murthy, K.R.; Goel, R.; Subbannayya, Y.; Jacob, H.K.C.; Murthy, P.R.; Manda, S.S.; Patil, A.H.; Sharma, R.; Sahasrabuddhe, N.A.; Parashar, A.; et al. Proteomic Analysis of Human Vitreous Humor. Clin. Proteom. 2014, 11, 29.
- Babizhayev, M.A.; Burke, L.; Micans, P.; Richer, S.P. N-Acetylcarnosine Sustained Drug Delivery Eye Drops to Control the Signs of Ageless Vision: Glare Sensitivity, Cataract Amelioration and Quality of Vision Currently Available Treatment for the Challenging 50,000-Patient Population. Clin. Interv. Aging 2009, 4, 31–50.
- Peynshaert, K.; Devoldere, J.; Minnaert, A.K.; De Smedt, S.C.; Remaut, K. Morphology and Composition of the Inner Limiting Membrane: Species-Specific Variations and Relevance toward Drug Delivery Research. Curr. Eye Res. 2019, 44, 465–475.
- Boye, S.L.; Bennett, A.; Scalabrino, M.L.; McCullough, K.T.; Van Vliet, K.; Choudhury, S.; Ruan, Q.; Peterson, J.; Agbandje-McKenna, M.; Boye, S.E. Impact of Heparan Sulfate Binding on Transduction of Retina by Recombinant Adeno-Associated Virus Vectors. J. Virol. 2016, 90, 4215–4231.
- Bisht, R.; Rupenthal, I.D.; Sreebhavan, S.; Jaiswal, J.K. Development of a Novel Stability Indicating RP-HPLC Method for Quantification of Connexin43 Mimetic Peptide and Determination of Its Degradation Kinetics in Biological Fluids. J. Pharm. Anal. 2017, 7, 365–373.
- Stampfli, H.F.; Quon, C.Y. Polymorphic metabolism of flestolol and other ester containing compounds by a carboxylesterase in New Zealand white rabbit blood and cornea. Res. Commun. Mol. Pathol. Pharmacol. 1995, 88, 87–97.
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug Discov. Today 2015, 20, 122–128.
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451.
- Radhakrishnan, K.; Sonali, N.; Moreno, M.; Nirmal, J.; Fernandez, A.A.; Venkatraman, S.; Agrawal, R. Protein delivery to the back of the eye: Barriers, carriers and stability of anti-VEGF proteins. Drug Discov. Today. 2017, 22, 416–423.
- Vaughan-Thomas, A.; Gilbert, S.J.; Duance, V.C. Elevated levels of proteolytic enzymes in the aging human vitreous. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3299–3304.
- Pescosolido, N.; Barbato, A.; Pascarella, A.; Giannotti, R.; Genzano, M.; Nebbioso, M. Role of Protease-Inhibitors in Ocular Diseases. Molecules 2014, 19, 20557–20569.
- Jwala, J.; Boddu, S.H.S.; Shah, S.; Sirimulla, S.; Pal, D.; Mitra, A.K. Ocular Sustained Release Nanoparticles Containing Stereoiso-meric Dipeptide Prodrugs of Acyclovir. J. Ocul. Pharmacol. Ther. 2011, 27, 163–172.
- Brinckerhoff, L.H.; Kalashnikov, V.V.; Thompson, L.W.; Yamshchikov, G.V.; Pierce, R.A.; Galavotti, H.S.; Engelhard, V.H.; Slingluff, C.L. Terminal Modifications Inhibit Proteolytic Degradation of an Immunogenic MART-127-35 Peptide: Implications for Peptide Vaccines. Int. J. Cancer 1999, 83, 326–334.
- Werle, M.; Bernkop-Schnürch, A. Strategies to Improve Plasma Half Life Time of Peptide and Protein Drugs. Amino Acids 2006, 30, 351–367.
- Attar, M.; Shen, J.; Ling, K.H.J.; Tang-Liu, D. Ophthalmic Drug Delivery Considerations at the Cellular Level: Drug-Metabolising Enzymes and Transporters. Expert Opin. Drug Deliv. 2005, 2, 891–908.
- Schwartzman, M.L.; Masferrer, J.; Dunn, M.W.; Mcgiff, J.C.; Abraham, N.G. Cytochrome P450, Drug Metabolizing Enzymes and Arachidonic Acid Metabolism in Bovine Ocular Tissues. Curr. Eye Res. 1987, 6, 623–630.
- Hayasaka, S. Lysosomal Enzymes in Ocular Tissues and Diseases. Surv. Ophthalmol. 1983, 27, 245–258.
- Vandervoort, J.; Ludwig, A. Ocular Drug Delivery: Nanomedicine Applications. Nanomedicine 2007, 2, 11–21.
- Ferrara, N.; Adamis, A. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug. Discov. 2016, 15, 385–403.
- Traynor, K. Aflibercept approved for macular degeneration. Am. J. Health Sys. Pharm. 2012, 69, 6.
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132.
- Theodossiadis, P.G.; Markomichelakis, N.N.; Sfikakis, P.P. Tumor necrosis factor antagonists: Preliminary evidence for an emerging approach in the treatment of ocular inflammation. Retina 2007, 27, 399–413.
- Harooni, M.; Freilich, J.M.; Abelson, M.; Refojo, M. Efficacy of hyaluronidase in reducing increases in intraocular pressure related to the use of viscoelastic substances. Arch. Ophthalmol. 1998, 116, 1218–1221.
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: Their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839.