Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Neuroblastoma is the most common extracranial pediatric tumor. Although children with low- and intermediate-risk neuroblastoma, which correspond to approximately half of all newly diagnosed cases, have a good event-free and overall survival, high-risk neuroblastoma can be extremely aggressive and hard-to-treat tumors. In neuroblastoma, p53 and TAp73 act as safeguards against malignant transformation, but they are commonly inhibited by negative regulators, such as MDMs, Itch, and Aurora kinase A.
- neuroblastoma
- p53 family proteins
- N-MYC
- miRNAs
1. Introduction
Neuroblastoma (NB) is a pediatric solid cancer with high prevalence in children younger than 10 years. It is considered one of the most frequent childhood tumors, accounting for 6–10% of all pediatric malignancies [1,2,3]. NB arises anywhere along the developing sympathetic nervous system from neural crest cells or their derivatives, such as Schwann cell precursors [4,5,6]. These cells normally migrate from the dorsal tube and differentiate to tissues or organs of the sympathetic nervous system; when this process fails, NB can be developed. For this reason, this solid tumor tends to appear in regions of the sympathetic nervous system, mainly in the abdomen and adrenal gland [7].
The heterogeneity of NB, which is reflected by several chromosomal aberrations, is considered a hallmark of this disease. This makes its treatment very challenging, especially due to frequent inter- and intra-tumorigenic heterogeneity in patients and the accumulation of gene mutations in recurrent tumor tissues [8]. Targeted therapy is being studied as a promising approach for the treatment of NB, particularly in high-risk patients.
One of the most known genetic factors for the development of high-risk NB is MYCN amplification [9,10,11]. Indeed, aberrant overexpression of the MYCN oncogene is associated with poor prognosis, tumor aggressiveness, and resistance to chemotherapy [9,10,11]. Another well-known genetic factor in NB is the anaplastic lymphoma kinase (ALK) mutation or amplification [12]. Most of the otherwise rare familial NB tumors (representing 1% of all NB cases) are associated with ALK germline mutations [13,14]. The resulting aberrant activity of ALK contributes to cell growth and survival of cancer cells by induction of pathways such as the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) and RAS/mitogen activated protein kinase (MAPK) [15]. In fact, over 10% of MYCN-amplified tumors bear ALK mutation [6,10]. The paired-like homeobox 2B (PHOX2B) is a transcription factor involved in the regulation of differentiation in the sympathetic nervous system [16]. Germline mutations of the PHOX2B gene, which result in a loss-of-function protein, predispose to NB development [17,18]. The loss-of-function of the RNA-helicase ATRX by a structural variant is another mechanism involved in the development of NB. These alterations commonly appear in older patients, and they are usually mutually exclusive of MYCN amplification [7,10]. Furthermore, in patients with poor outcome and high-risk NB, the telomerase reverse transcriptase (TERT) is activated by genetic rearrangements [19]. Once activated, telomere lengthening occurs, which might be correlated to the aggressiveness of some types of NB [19]. NB prognosis may also be associated with altered expression of tropomyosin receptor kinase (Trk) proteins. In particular, TrkB is overexpressed in NB cases with amplified MYCN, being its activation related to increased proliferation, angiogenesis and chemoresistance of NB cells [20]. Despite this, higher expression of TrkA can also be found in types of NB diagnosed at early ages and without MYCN amplification, therefore being associated with better outcomes [10,11]. Some compounds targeting these pathways have already entered clinical trials.
Chromosome gain and loss are also related to NB development. In particular, gain of parts of the chromosome 17q and loss of chromosome 1p are associated with MYCN amplification in NB, as well as poor prognosis [7]. On the other hand, loss of 11q is inversely correlated with MYCN amplification. However, this chromosomal loss is also associated with poor prognosis in NB [21,22].
Loss of heterozygosity (LOH) of 1p36 is common in high-risk NB. Interestingly, the smallest deleted region shared across NB tumors comprises TP73, encoding the tumor suppressor protein TAp73 [23]. Indeed, LOH of TP73 was associated with MYCN amplification and subsequently a high-risk NB [24].
The p53 and TAp73 proteins function as molecular hubs of an intricated and robust carcinogenic signaling network, coordinating cell proliferation, death, and differentiation, among many other pivotal cellular processes [25].
2. The p53 Family Proteins: p53 and TAp73
2.1. p53 and TAp73 Interaction with MDM2 and MDMX
Several studies have demonstrated that murine double minute (MDM)2 has a pathogenic role in NB, and that targeting this regulator could be an interesting therapeutic approach. In addition, MDM2 overexpression in NB is more prevalent in relapsed cases rather than in primary NB, indicating a correlation between MDM2 overexpression and poor prognosis in NB [41].
NB commonly harbors a wild-type (wt)p53 form, which is inactivated by interaction with inhibitory proteins such as MDM2, and its homologue MDMX (reviewed in [27,42]) (Figure 1b). In fact, in many types of cancer, including NB [43], MDM2 and MDMX are overexpressed or amplified, acting as oncogenes, and leading to therapeutic resistance and metastasis (reviewed in [44,45,46,47]). The inhibition of TAp73 by these endogenous regulators has also been related to NB, especially in high-risk and relapsed cases [45].
By binding to p53 and TAp73, MDM2 and MDMX can inhibit their transcriptional activity. MDM2 can also bind to p53 and act as an E3 ubiquitin ligase, targeting p53 for proteasomal degradation. In fact, MDM2 mediates the nuclear export of p53 to the cytosol, where it can be mono- or polyubiquitinylated. It should be noted that MDM2 is transcriptionally induced by p53, forming a feedback loop [48,49]. Interestingly, conversely to previous data, a recent study demonstrated that TAp73 may also be degraded in vitro and in vivo, through polyubiquitination by MDM2, being Lys11, Lys29 and Lys63 residues the main targets of MDM2 for TAp73 degradation [50]. Although MDMX does not have ubiquitin ligase activity, it can form a heterodimer with MDM2, promoting this activity by MDM2 and being itself inhibited by MDM2 (reviewed in [42,47]) (Figure 1b). Unlike p53, TAp73 does not undergo MDM2-mediated nuclear export. Instead, TAp73 accumulates in the nucleus of MDM2-expressing cells as aggregates [51], suggesting a structural variation between p53 and TAp73 that differentiates this subcellular distribution. Importantly, MDM2 overexpression has been shown to promote drug resistance in wtp53-proficient NB cells via inactivating p53/TAp73, which results in the transcriptional downregulation of the pro-apoptotic protein NOXA [52].
Preclinical studies have evidenced that NB cells are very sensitive to inhibitors of MDM2, such as nutlin-3a [53,54,55] (Figure 2). Despite this, the number of MDM2 inhibitors under clinical trials for NB treatment is very limited, only comprising one ongoing study with Idasanutlin (also called RG7388) (Table 1). In fact, some concerns related to the use of these compounds, particularly the development of drug resistance and tumor relapse, have been reported in several studies and are closely associated with the appearance of mutated p53 forms upon long-term therapeutic exposures [45].
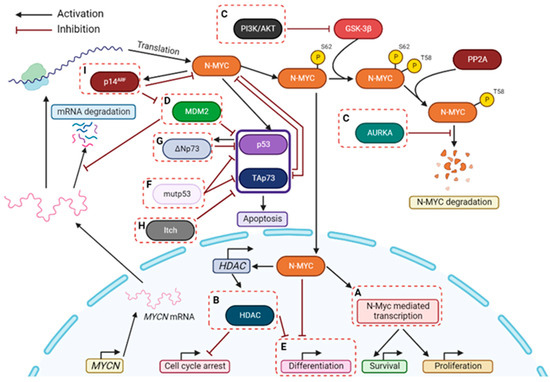
Figure 2. Therapeutic strategies for targeting NB, particularly with MYCN amplification. Some possible strategies to treat MYCN-amplified NB patients are highlighted in dashed boxes and may include: (A) Inhibition of N-MYC-dependent transcription with BET-bromodomain inhibitors; (B) Inhibition of HDACs; (C) Inhibition of proteins involved in stabilizing N-MYC; (D) Suppression of MDM2 (which stabilizes MYCN mRNA and disrupts p53-mediated apoptosis); (E) Induction of differentiation; (F) Destabilization of mutp53-TAp73 interaction; (G) Inhibition of ΔNp73; (H) Inhibition of Itch; (I) Activation of p14ARF. P: Phosphorylation.
An important regulator of the p53-MDM2 network is the tumor suppressor p14ARF. By forming a complex with MDM2, p14ARF inhibits MDM2-dependent p53 degradation and maintains p53 in an active state [56] (Figure 1b). In fact, deletion or downregulation of the INK4a-ARF gene (encoding p14ARF) have represented a major mechanism of p53 inactivation in NB [57]. Consistently, the loss of p14ARF has been observed in relapse NB tumors [58]. Interestingly, p14ARF can also exhibit p53-independent tumor suppressor activity by directly binding and inhibiting c-MYC and N-MYC [52] (Figure 1b). Stimulation of p14ARF would therefore represent an additional therapeutic strategy against NB (Figure 2).
2.2. p53 and TAp73 Interaction with Mutant p53
Most p53 mutations occur in the p53 DBD via single amino acid substitutions (missense mutations), which are translated into loss-of-function of the tumor suppressor capacity, or in some cases into oncogenic gain-of-function (GOF) (reviewed in [59]). Mutations in this region also lead to cellular accumulation of mutant (mut)p53, since MDM2 cannot recognize mutp53 and initiate ubiquitin-mediated degradation (reviewed in [59]). Cancers with mutp53 are also associated with increased proliferation, metastasis, angiogenesis, genomic instability, and resistance to therapy [59,60].
Mutp53 is an inhibitor of various transcription factors, including wtp53 [61,62] and TAp73 [63,64] (Figure 1b). It is thought that GOF activity of mutp53 depends on these interactions to mediate the transcription of several genes [65]. Besides affecting transcription factors, mutp53 can also promote the stability of microRNAs (miRNAs) involved in tumor progression and dissemination [66,67,68] (see Section 4).
The TAp73 inhibition by mutp53 is highly related to high-risk and relapsed cancers [69]. In fact, the binding of mutp53 to TAp73 DBD results in the inhibition of TAp73-dependent transactivation and apoptosis [63], and promotion of drug resistance [70,71].
Consistently, even though p53 mutations are rare at diagnosis in NB (<2% in primary tumors and around 15% in relapsed ones) [72,73], various reports have shown that they are associated with the development of drug resistance [74,75]. In line with this, most of the reported mutp53-related NB clinical cases are associated with relapse and poor outcome [72,76,77]. It has also been shown that cytotoxic therapy for NB, including doxorubicin, cisplatin, and vincristine, can induce p53 mutations. In fact, NB cells derived from the same patient before (SK-N-BE(1)) and after (SK-N-BE(2)) chemotherapy, showed alterations in the p53 status (from wt to mut) and therapeutic response (from chemosensitive to chemoresistant) [78]. Indeed, acquired drug resistance has been a major barrier to the successful treatment of many cancers, including NB [79].
Hence, disruption of the mutp53 interaction with transcription factors such as p53 and TAp73 reveals great therapeutic potential against mutp53-profecient NB tumors (Figure 2). In fact, the treatment of cancer cells harboring mutp53 with RETRA, a destabilizer of the mutp53-TAp73 interaction, significantly increased TAp73 expression levels and subsequent transcriptional activity [80,81]. More recently, the xanthone derivative 3,4-dimethoxy-9-oxo-9H-xanthene-1-carbaldehyde (LEM2) was uncovered as a potent anticancer agent against patient-derived NB cells [82]. LEM2 inhibited both TAp73-mutp53 and TAp73-MDM2 interactions, resulting in TAp73 activation and induction of cell cycle arrest and apoptosis in NB cells. In these cells, LEM2 also showed promising synergistic effects with chemotherapeutics such as doxorubicin and cisplatin [82].
2.3. p53 and TAp73 Interaction with ΔNp73
As previously mentioned, ∆Np73 acts as an oncogene that is associated with cancer development, metastasis, and drug resistance [30,83]. In fact, in 2002, Casciano et al., showed that expression of ∆Np73 was related to decreased apoptosis in vivo, being a robust predictor of unfavorable outcome, regardless of age, primary tumor site, stage, chromosome 1p deletion, and amplification of MYCN in NB [84]. In NB, ∆Np73 is considered a poor prognosis marker, namely due to its interaction with p53 and TAp73 and subsequent inhibition of their transcriptional activity (reviewed in [85,86]).
In fact, the effect of TAp73 in NB is thought to depend on the ratio between TAp73 and ΔNp73 isoforms. Several mechanisms responsible for deregulating this ratio have been described in NB, including hyper- or hypomethylation, which are crucial events in cell transformation [87]. Since some human malignancies, such as non-Hodgkin lymphoma, display TP73 silencing due to promoter methylation [88], it was suggested that this type of modification could also account for the decreased levels of TAp73 in NB. However, this idea gained less support based on the impossibility of establishing a correlation between the TP73 promoter methylation status and the TAp73 expression in NB [89].
Unlike TAp73, ΔNp73 has been reported to be overexpressed in primary NB [84]. These increased ΔNp73 levels might be responsible for the inhibition of the pro-apoptotic activity of p53 [90] and may even block the TAp73 activity, allowing neural cells to escape from the TAp73-mediated differentiation process [36]. ΔNp73 is also capable of inhibiting the activation of ATM and p53, making NB more resistant to chemotherapeutics [91]. From a mechanistic point of view, this overexpression of ΔNp73 might be related to epigenetic modifications, such as hypomethylation of the internal P2 promoter, which controls the transcription of this isoform and has already been observed in NB cells and primary tumors [89,92].
It has become evident that ∆Np73 has high clinical significance as a marker for NB severity [85]. ∆Np73 is not just a relative of p53, but has created its own identity, also becoming an encouraging target for NB therapy (Figure 2).
2.4. Proteasomal-Dependent Degradation of TAp73 by Itch
The E3 ubiquitin ligases (E3s) have proven to play a fundamental role in the regulation of cell proliferation, differentiation, and apoptosis. Consistently, genetic alterations and dysfunctions in the E3s activity have been deeply related to tumor progression [93,94]. The HECT-type E3 ubiquitin ligase Itch has been reported to regulate apoptosis, cell growth and inflammation pathways, and some studies have even shown that its dysregulated expression interfered with the apoptotic response induced by conventional chemotherapy [93,94,95]. In fact, the depletion of Itch by siRNA has sensitized lung cancer cells to anti-proliferative effects of gemcitabine [96]. Similarly, RNA interference-mediated downregulation of Itch significantly enhanced suppression of pancreatic cancer growth by gemcitabine in vivo [97]. This was further demonstrated in NB cells by Meng et al., who used in vivo nano-delivery of the Itch siRNA in NB xenograft mouse models to sensitize tumor cells to radiotherapy [98]. Itch is responsible for the regulation of the proteasomal-dependent degradation of a group of target proteins, including TAp73 [99] (Figure 1b). Interestingly, from the several E3s that control TAp73 protein levels [100,101,102], Itch is one of the most characterized. Specifically, it stimulates the proteasome-dependent degradation of TAp73 in unstressed cells, keeping its expression levels low in normal conditions [103]. As shown in many cancer cell lines, in response to chemotherapeutic drugs, the induction of TAp73 activity seems to be, at least in part, accomplished through downregulation of Itch [99].
Since most NB cell lines express Itch, it may be possible that TAp73 levels are negatively controlled by an Itch-dependent mechanism, which could explain the chemoresistance in NB [9]. As such, targeting Itch could represent a strategy of TAp73 stabilization, thus enhancing pro-apoptotic activity of TAp73 and even sensitizing NB cells to commonly used chemotherapeutic drugs. In 2014, Rossi et al. identified desmethylclomipramine, the active metabolite of clomipramine, as an inhibitor of Itch autoubiquitylation activity and Itch-dependent ubiquitylation of TAp73 [104]. Clomipramine is an FDA-approved drug used in the treatment of obsessive-compulsive disorders [105]. Interestingly, this drug also increases the cytotoxic activity of conventional chemotherapeutics in cancer cell lines and cancer stem cells [96,104]. Although it is still unclear if this effect is completely dependent on Itch inhibition, these data suggest that targeting Itch could represent a novel therapeutic approach for NB treatment (Figure 2).
2.5. p53 and TAp73 Interaction with AURKA
Aurora kinases are a family of serine/threonine protein kinases that have a crucial role in cellular division. These kinases are essential to ensure the correct replication of the genetic information, as well as for the maintenance of genomic and chromosomal integrity during cell division [106]. The aurora A (AURKA) is the most studied member of Aurora kinase family, mainly due to its central role in mitotic regulation and high expression levels in many types of cancers, including NB [107]. Most of the AURKA proteins are activated in late G2 phase, and their activity is maintained until the end of mitosis [108]. In mitosis, AURKA is mainly involved in centrosome maturation, mitotic entry regulation, and spindle assembly. Once mitosis is completed, most AURKA proteins are degraded, and only a small amount is detected in G1 phase [109].
AURKA is known to acquire gain-of-function alterations, mainly due to amplification, overexpression of its gene and p53 loss-of-function. These events have been associated with several cellular phenotypes, such as centrosome amplification, override of spindle assembly, and DNA damage checkpoint response and aneuploidy [110]. The induction of these phenotypes suggests that AURKA and p53 are involved in overlapping signaling pathways that are responsible for the regulation of these aberrant cellular outcomes. This was first proven in 2002 by Marumoto et al., who demonstrated that p53 was able to suppress the oncogenic effects of AURKA through physiological interaction, in a transactivation-independent manner [111] (Figure 1b). Additionally, p53 has been shown to downregulate AURKA expression, as well as its kinase activity and stability, by binding to AURKA promoter or through activation of p53 target genes, such as CDKN1A (encoding p21), GADD45A and FBXW7α. The induction of p21 inhibits Cdk kinase activity, which leads to the maintenance of RB1 in a hypophosphorylated state in a complex with E2F3. This impairs the AURKA gene expression [110]. On the other hand, GADD45 inhibits AURKA kinase activity through direct interaction, preventing cells from centrosome amplification and aborted cytokinesis [112]. These results suggest that the inhibition of AURKA by p53 is important for the maintenance of centrosome number and chromosomal and genomic stability. Regarding the tumor suppressor protein FBXW7α, this p53-dependent protein is a component of the SCF-like ubiquitin ligase complex that targets both AURKA and AURKB for proteasomal degradation [113,114,115]. FBXW7α is frequently downregulated or mutated in tumors. It cooperates with PTEN in the regulation of AURKA degradation via the PI3K/AKT/GSK3β pathway, mainly participating in the degradation of active AURKA proteins [114,116]. The dysfunction of the p53-FBXW7α axis is frequently observed in human tumors, and it has been proven that this deregulation can mediate AURKA-induced centrosome amplification, leading to aneuploidy [114,117]. It should also be mentioned that AURKA is able to stabilize N-MYC by interfering with its FBXW7α-mediated degradation, as demonstrated by a synthetic lethal screening of proteins interacting with N-MYC [118]. Even though it was reported that this interaction is independent of AURKA kinase activity, a study by Brockmann et al. demonstrated that inhibitors of AURKA kinase activity could also disrupt the interaction between AURKA and FBXW7α, leading to N-MYC destabilization and tumor regression in a mouse model of N-MYC-driven NB xenograft [119].
Some studies have reported that Aurora kinases negatively regulate p53 through phosphorylation-mediated posttranslational modification of either p53 itself or of interactor proteins that bind to p53, which may result in failure of the DNA damage checkpoint, as well as lack of response to cell death induction in AURKA overexpressing cells [110]. In fact, AURKA phosphorylates p53 at serine 315, which facilitates MDM2-mediated ubiquitination and degradation of p53 [120]. Furthermore, AURKA phosphorylation of p53 at serine 215 inhibits p53 DNA-binding and subsequent transactivation activity [121]. The role of AURKA in TAp73 regulation was first demonstrated, in 2008, by Dar et al., who showed that the treatment with the AURKA inhibitor MLN8054 or knockdown of AURKA, in p53-deficient cells, induced TAp73-mediated apoptosis [122]. Later, in 2012, AURKA was discovered to directly interact with and phosphorylate TAp73 DBD at serine 235, which resulted in the loss of DNA-binding ability and transactivation activity of TAp73 [123]. These events were observed in cells becoming resistant to DNA damage-induced cell death [123].
It has been demonstrated that spindle assembly checkpoint (SAC) override is associated with aberrant AURKA expression, regardless of p53 status in cells [110]. As a result, it is currently unknown if p53 plays a role in AURKA signaling for SAC override. There is some evidence suggesting that p53 is also involved in mitotic cell death and postmitotic checkpoint following aberrant mitosis or spindle damage by interacting with SAC proteins, rather than activating SAC [124,125,126,127]. However, the role of TAp73 interaction with AURKA in SAC is better understood and defined. Some in vitro studies describe a role for TAp73 in G2-M transition, mitotic exit, and mitotic cell death [124,125,126,127], while an in vivo study in transgenic mice lacking TAp73 has suggested that the frequent occurrence of aberrant spindle structure is associated with aneuploidy and chromosome instability [128]. Additionally, biochemical studies have shown that TAp73 interacted with the SAC proteins BUB1, BUB3 and BUBR1, which are crucial for BUB1 and BUBR1 localization at kinetochores and BUBR1 kinase activity [128,129]. These data indicated that TAp73 was directly involved in regulating SAC signaling to maintain chromosomal stability. These results have suggested that AURKA-TAp73 interaction was crucial for a critical step in the SAC inactivation pathway. However, unlike its effect on the MAD2-CDC20 interaction, phosphorylation of TAp73 did not affect the interaction of BUBR1 with CDC20 and its kinetochore localization, which indicated that TAp73 had a role in a distinct pathway to control SAC activation [129].
Cancer cells with ectopic expression of ΔNp73 show abnormal mitotic progression, followed by multipolar spindle and cytokinesis failure, which results in multinucleated cells [110]. However, ΔNp73 seems not to affect either SAC activation in the presence of spindle poison, or interaction with BUBR1 [128,130], suggesting a contribution of ΔNp73 to bypassing SAC. Interestingly, AURKA also interacts with and phosphorylates ΔNp73, but its phosphorylation site seems to be different from TAp73 and remains to be mapped [123]. The role played by this interaction has not yet been elucidated.
In 2021, Yi et al. showed how inhibitors of AURKA, such as alisertib, were highly synergistic with BET bromodomain inhibitors, in NB cells [131]. Consistently, they observed a decreased MYCN mRNA levels due to BET bromodomain inhibitors, which downregulate the transcription of MYCN, and increased degradation of N-MYC through AURKA inhibitors. They further evidenced the induction of apoptosis and cell cycle arrest in response to this combination, mainly in a context of a functional TP53. Interestingly, the results indicated that TP53 status may be predictive of therapeutic response to AURKAi, in NB cells, since TP53 loss conferred resistance to alisertib monotherapy. These data therefore reinforce the beneficial effect that activators of the p53 pathway may have in combination with AURKAi. More recently, a study from Nguyen et al. [132] found that selinexor, an inhibitor of the nuclear export protein XPO1, induced p53 phosphorylation at serine 315, an initiating step for p53 degradation undertaken by AURKA, as previously referred. By using alisertib, p53-mediated cell death was enhanced in NB xenograft mouse models.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14246212
This entry is offline, you can click here to edit this entry!