Bortezomib is the most effective chemotherapeutic drug used in the treatment of MM. This inhibitor is a dipeptide boronic acid analogue discovered in 1995 and is the premier in the class of chymotrypsin-like (CP) inhibitors. Bortezomib is a C-terminal boronic acid, and the boron atom is essential for inhibiting the proteasome activity because of its ability to specifically and tightly bind the β5 catalytic subunit. Boronates form tetrahedral adducts, which are further stabilized by a hydrogen bond between the N-terminal amino group of threonines and the hydroxyl groups of boronic acid. These bonds provide a higher influence for Bortezomib than other drugs developed for inhibition. It binds the proteasome with a high resolution, slowly dissociates, and provides a stable but reversible proteasome inhibition. Various mechanisms have been suggested to explain the multidrug resistance in cancer cells. Increased drug excretion, decreased drug uptake, activation of detoxification systems, inhibition of apoptosis, alterations in cell cycle regulation factors, and changes in drug targets are among the causes.
- multiple myeloma
- Bortezomib
- multidrug resistance
- nanoparticle
1. Abnormal Drug Transport
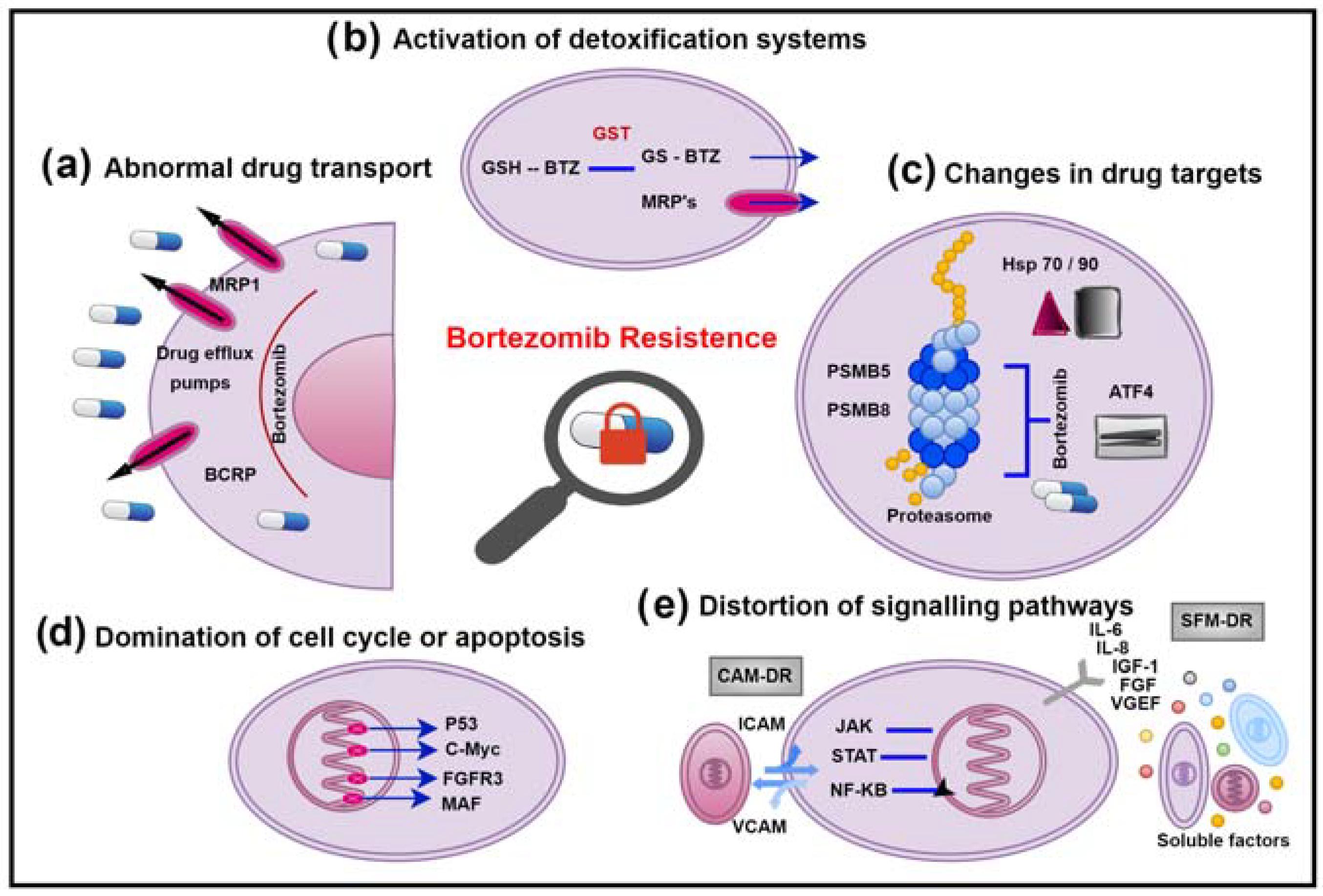
2. Activation of Detoxification Systems
3. Changes in Drug Targets
4. Domination of Cell Cycle or Apoptosis
5. Distortion of Signalling Pathways
This entry is adapted from the peer-reviewed paper 10.3390/ph16010111
References
- Ahmed, S.; Khan, H.; Aschner, M.; Mirzae, H.; Küpeli Akkol, E.; Capasso, R. Anticancer Potential of Furanocoumarins: Mechanistic and Therapeutic Aspects. Int. J. Mol. Sci. 2020, 21, 5622.
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464.
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121.
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys Acta 1976, 455, 152–162.
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295.
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 26, 1–9.
- Kozalak, G.; Oksuzoglu, E. Efficacy of Multi-Drug Resistance Transporters and Glutathione S-Transferase P-1 at Developing Bortezomib Resistance in Multiple Myeloma Cell Lines. Lat. Am. J. Pharm. 2021, 40, 2709–2716.
- Grogan, T.M.; Spier, C.M.; Salmon, S.E.; Matzner, M.; Rybski, J.; Weinstein, R.S.; Scheper, R.J.; Dalton, W.S. P-glycoprotein expression in human plasma cell myeloma: Correlation with prior chemotherapy. Blood 1993, 81, 490–495.
- Verbrugge, S.E.; Assaraf, Y.G.; Dijkmans, B.A.; Scheffer, G.L.; Al, M.; den Uyl, D.; Oerlemans, R.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; et al. Inactivating PSMB5 Mutations and P-Glycoprotein (Multidrug Resistance-Associated Protein/ATP-Binding Cassette B1) Mediate Resistance to Proteasome Inhibitors: Ex Vivo Efficacy of (Immuno)Proteasome Inhibitors in Mononuclear Blood Cells from Patients with Rheumatoid Arthritis. J. Pharmacol. Exp. Ther. 2012, 341, 174–182.
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357–1368.
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670.
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van De Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464.
- Mo, W.; Zhang, J.T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27.
- Chernykh, Y.; Golenkov, A.; Vysotskaya, L.; Shushanov, S.; Rybalkina, E. Effect of Expression of Multidrug Resistance Genes in Newly Diagnosed Multiple Myeloma on the Clinical Course of the Disease. Blood 2016, 128, 5144.
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889.
- Scheffer, G.L.; Wijngaard, P.L.J.; Flens, M.J.; Izquierdo, M.A.; Slovak, M.L.; Pinedo, H.M.; Meijer, C.J.L.M.; Clevers, H.C.; Scheper, R.J. The drug resistance-related protein LRP is the human major vault protein. Nat. Med. 1995, 1, 578–582.
- Krishnan, S.R.; Jaiswal, R.; Brown, R.D.; Luk, F.; Bebawy, M. Multiple myeloma and persistence of drug resistance in the age of novel drugs (Review). Int. J. Oncol. 2016, 49, 33–50.
- Kulsoom, B.; Shamsi, T.S.; Afsar, N.A. Lung resistance-related protein (LRP) predicts favorable therapeutic outcome in Acute Myeloid Leukemia. Sci. Rep. 2019, 9, 378.
- Huh, H.J.; Park, C.-J.; Jang, S.; Seo, E.-J.; Chi, H.-S.; Lee, J.-H.; Lee, K.-H.; Seo, J.J.; Moon, H.N.; Ghim, T. Prognostic Significance of Multidrug Resistance Gene 1 (MDR1), Multidrug Resistance-related Protein (MRP) and Lung Resistance Protein (LRP) mRNA Expression in Acute Leukemia. J. Korean Med. Sci. 2006, 21, 253–258.
- Mándoky, L.; Géczi, L.; Doleschall, Z.; Bodrogi, I.; Csuka, O.; Kásler, M.; Bak, M. Expression and prognostic value of the lung resistance-related protein (LRP) in germ cell testicular tumors. Anticancer Res. 2004, 24, 1097–1104.
- Burger, H.; Foekens, J.A.; Look, M.P.; Meijer-van Gelder, M.E.; Klijn, J.G.; Wiemer, E.A.; Stoter, G.; Nooter, K. RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: Correlation with chemotherapeutic response. Clin. Cancer Res. 2003, 9, 827–836.
- Raaijmakers, H.G.; Izquierdo, M.A.; Lokhorst, H.M.; de Leeuw, C.; Belien, J.A.; Bloem, A.C.; Dekker, A.W.; Scheper, R.J.; Sonneveld, P. Lung-resistance-related protein expression is a negative predictive factor for response to conventional low but not to intensified dose alkylating chemotherapy in multiple myeloma. Blood 1998, 91, 1029–1036.
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.V.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654.
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085.e1079.
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291.
- Loe, D.W.; Deeley, R.G.; Cole, S.P.C. Characterization of Vincristine Transport by the Mr 190,000 Multidrug Resistance Protein (MRP): Evidence for Cotransport with Reduced Glutathione1. Cancer Res. 1998, 58, 5130–5136.
- Renes, J.; de Vries, E.G.; Nienhuis, E.F.; Jansen, P.L.; Müller, M. ATP- and glutathione-dependent transport of chemotherapeutic drugs by the multidrug resistance protein MRP1. Br. J. Pharmacol. 1999, 126, 681–688.
- Nasr, R.; Lorendeau, D.; Khonkarn, R.; Dury, L.; Pérès, B.; Boumendjel, A.; Cortay, J.C.; Falson, P.; Chaptal, V.; Baubichon-Cortay, H. Molecular analysis of the massive GSH transport mechanism mediated by the human Multidrug Resistant Protein 1/ABCC1. Sci. Rep. 2020, 10, 7616.
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013, 23, 48–62.
- Ishikawa, T.; Müller, M.; Klünemann, C.; Schaub, T.; Keppler, D. ATP-dependent primary active transport of cysteinyl leukotrienes across liver canalicular membrane. Role of the ATP-dependent transport system for glutathione S-conjugates. J. Biol. Chem. 1990, 265, 19279–19286.
- Lagas, J.S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Hepatic Clearance of Reactive Glucuronide Metabolites of Diclofenac in the Mouse Is Dependent on Multiple ATP-Binding Cassette Efflux Transporters. Mol. Pharmacol. 2010, 77, 687–694.
- Kool, M.; van der Linden, M.; de Haas, M.; Baas, F.; Borst, P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 1999, 59, 175–182.
- Hopper-Borge, E.; Xu, X.; Shen, T.; Shi, Z.; Chen, Z.-S.; Kruh, G.D. Human Multidrug Resistance Protein 7 (ABCC10) Is a Resistance Factor for Nucleoside Analogues and Epothilone B. Cancer Res. 2008, 69, 178–184.
- Guo, Y.; Köck, K.; Ritter, C.A.; Chen, Z.-S.; Grube, M.; Jedlitschky, G.; Illmer, T.; Ayres, M.; Beck, J.F.; Siegmund, W.; et al. Expression of ABCC-Type Nucleotide Exporters in Blasts of Adult Acute Myeloid Leukemia: Relation to Long-term Survival. Clin. Cancer Res. 2009, 15, 1762–1769.
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236.
- Petrini, M.; Di Simone, D.; Favati, A.; Mattii, L.; Valentini, P.; Grassi, B. GST-pi and P-170 co-expression in multiple myeloma. Br. J. Haematol. 1995, 90, 393–397.
- Ishikawa, T. The ATP-dependent glutathione S-conjugate export pump. Trends Biochem. Sci. 1992, 17, 463–468.
- Zaman, G.J.; Lankelma, J.; Tellingen, O.V.; Beijnen, J.; Dekker, H.; Paulusma, C.; Elferink, R.P.O.; Baas, F.; Borst, P. Role of glutathione in the export of compounds from cells by the multidrug-resistance-associated protein. Proc. Natl. Acad. Sci. USA 1995, 92, 7690–7694.
- Keppler, D.; König, J. Expression and localization of the conjugate export pump encoded by the MRP2 (cMRP/cMOAJ) gene in liver. FASEB J. 1997, 11, 509–515.
- Armstrong, R.N. Enzyme-catalyzed detoxication reactions: Mechanisms and stereochemistry. CRC Crit. Rev. Biochem. 1987, 22, 39–88.
- Sau, A.; Pellizzari Tregno, F.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122.
- Tew, K.D.; Dutta, S.; Schultz, M. Inhibitors of glutathione S-transferases as therapeutic agents. Adv. Drug Deliv. Rev. 1997, 26, 91–104.
- Morrow, C.S.; Smitherman, P.K.; Townsend, A.J. Combined expression of multidrug resistance protein (MRP) and glutathione S-transferase P1-1 (GSTP1-1) in MCF7 cells and high level resistance to the cytotoxicities of ethacrynic acid but not oxazaphosphorines or cisplatin. Biochem. Pharmacol. 1998, 56, 1013–1021.
- Zhao, J.; Wang, M.; He, P.; Chen, Y.; Wang, X.; Zhang, M. Identification of glutathione S-transferase π 1 as a prognostic proteomic biomarker for multiple myeloma using proteomic profiling. Oncol. Lett. 2020, 19, 2153–2162.
- Holkova, B.; Grant, S. Proteasome inhibitors in mantle cell lymphoma. Best Pract. Res. Clin. Haematol. 2012, 25, 133–141.
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916.
- Egan, P.; Drain, S.; Conway, C.; Bjourson, A.J.; Alexander, H.D. Towards Stratified Medicine in Plasma Cell Myeloma. Int. J. Mol. Sci. 2016, 17, 1760.
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207.
- Nikesitch, N.; Ling, S.C. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97–101.
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104.
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666.
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423.
- Manik, C.; Mindaugas, A.; Thorsten, S.; Elisabeth, M.; Claudia, H.; Torsten, S.; Tanja, H.; Heike, S.; Stefanie, K.; Hermann, E.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141.
- Huber, E.M.; Heinemeyer, W.; Groll, M. Bortezomib-resistant mutant proteasomes: Structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015, 23, 407–417.
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768.
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961.
- Johnsen, A.; France, J.; Sy, M.S.; Harding, C.V. Down-regulation of the transporter for antigen presentation, proteasome subunits, and class I major histocompatibility complex in tumor cell lines. Cancer Res. 1998, 58, 3660–3667.
- Suzuki, E.; Demo, S.; Deu, E.; Keats, J.; Arastu-Kapur, S.; Bergsagel, P.L.; Bennett, M.K.; Kirk, C.J. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS ONE 2011, 6, e27996.
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160.
- Furukawa, Y.; Kikuchi, J. Molecular basis of clonal evolution in multiple myeloma. Int. J. Hematol. 2020, 111, 496–511.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511.
- Öksüzoğlu, E.; Kozalak, G. Inhibition of apoptosis may lead to the development of bortezomib resistance in multiple myeloma cancer cells. Turk. J. Biochem. 2021, 46, 65–71.
- Sekiguchi, N.; Ootsubo, K.; Wagatsuma, M.; Midorikawa, K.; Nagata, A.; Noto, S.; Yamada, K.; Takezako, N. The impact of C-Myc gene-related aberrations in newly diagnosed myeloma with bortezomib/dexamethasone therapy. Int. J. Hematol. 2014, 99, 288–295.
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208.
- Solary, E.; Droin, N.; Bettaieb, A.; Corcos, L.; Dimanche-Boitrel, M.T.; Garrido, C. Positive and negative regulation of apoptotic pathways by cytotoxic agents in hematological malignancies. Leukemia 2000, 14, 1833–1849.
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207.
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379.
- Spets, H.; Strömberg, T.; Georgii-Hemming, P.; Siljason, J.; Nilsson, K.; Jernberg-Wiklund, H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: Regulation during interleukin-6(IL-6)-induced growth and survival. Eur. J. Haematol. 2002, 69, 76–89.
- Balsas, P.; López-Royuela, N.; Galán-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812.
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN- and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304.
- Xie, T.; Geng, J.; Wang, Y.; Wang, L.; Huang, M.; Chen, J.; Zhang, K.; Xue, L.; Liu, X.; Mao, X.; et al. FOXM1 evokes 5-fluorouracil resistance in colorectal cancer depending on ABCC10. Oncotarget 2017, 8, 8574–8589.
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375.
- Bi, C.; Chng, W.J. MicroRNA: Important Player in the Pathobiology of Multiple Myeloma. BioMed Res. Int. 2014, 2014, 521586.
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890.
- Chen, L.; Li, C.; Zhang, R.; Gao, X.; Qu, X.; Zhao, M.; Qiao, C.; Xu, J.; Li, J. miR-17-92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett. 2011, 309, 62–70.
- Zhang, Y.K.; Wang, H.; Leng, Y.; Li, Z.L.; Yang, Y.F.; Xiao, F.J.; Li, Q.F.; Chen, X.Q.; Wang, L.S. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem. Biophys. Res. Commun. 2011, 414, 233–239.
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333.
- Gururajan, M.; Haga, C.L.; Das, S.; Leu, C.M.; Hodson, D.; Josson, S.; Turner, M.; Cooper, M.D. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int. Immunol. 2010, 22, 583–592.
- Neri, P.; Gratton, K.; Ren, L.; Mansoor, A.; Duggan, P.; Stewart, D.A.; Bahlis, N.J. miRNA Expression in Multiple Myeloma as Predictive Model of Response to Bortezomib. Blood 2009, 114, 4918.
- Morelli, E.; Biamonte, L.; Federico, C.; Amodio, N.; Di Martino, M.T.; Gallo Cantafio, M.E.; Manzoni, M.; Scionti, F.; Samur, M.K.; Gullà, A.; et al. Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92. Blood 2018, 132, 1050–1063.
- Yang, W.C.; Lin, S.F. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. BioMed Res. Int. 2015, 2015, 341430.
- Chauhan, D.; Hideshima, T.; Anderson, K.C. Proteasome inhibition in multiple myeloma: Therapeutic implication. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 465–476.
- Cusack, J.C. Rationale for the treatment of solid tumors with the proteasome inhibitor bortezomib. Cancer Treat. Rev. 2003, 29 (Suppl. S1), 21–31.
- Fabre, C.; Mimura, N.; Bobb, K.; Kong, S.Y.; Gorgun, G.; Cirstea, D.; Hu, Y.; Minami, J.; Ohguchi, H.; Zhang, J.; et al. Dual inhibition of canonical and noncanonical NF-κB pathways demonstrates significant antitumor activities in multiple myeloma. Clin. Cancer Res. 2012, 18, 4669–4681.
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol. Cancer Res. 2008, 6, 1356–1364.
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674.
- Schmidmaier, R.; Mörsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for cell adhesion-mediated drug resistance of multiple myeloma cells in vivo. Int. J. Biol. Markers 2006, 21, 218–222.
- Cencini, E.; Fabbri, A.; Sicuranza, A.; Gozzetti, A.; Bocchia, M. The Role of Tumor-Associated Macrophages in Hematologic Malignancies. Cancers 2021, 13, 3597.
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol. Cancer 2010, 9, 176.
- Yang, Y.; Chen, Y.; Saha, M.N.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015, 29, 715–726.
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566.
- Yang, Y.; Shi, J.; Tolomelli, G.; Xu, H.; Xia, J.; Wang, H.; Zhou, W.; Zhou, Y.; Das, S.; Gu, Z.; et al. RARα2 expression confers myeloma stem cell features. Blood 2013, 122, 1437–1447.