Cluster of differentiation 36 (CD36) belongs to the B2 receptors of the scavenger receptor class B family, which is comprised of single-chain secondary transmembrane glycoproteins. It is present in a variety of cell types, including monocytes, macrophages, microvascular endothelial cells, adipocytes, hepatocytes, platelets, skeletal muscle cells, kidney cells, cardiomyocytes, taste bud cells, and a variety of other cell types. CD36 can be localized on the cell surface, mitochondria, endoplasmic reticulum, and endosomes, playing a role in lipid accumulation, oxidative stress injury, apoptosis, and inflammatory signaling. CD36 is expressed in a variety of ocular cells, including retinal pigment epithelium (RPE), retinal microvascular endothelial cells, retinal ganglion cells (RGC), Müller cells, and photoreceptor cells, playing an important role in eye diseases, such as age-related macular degeneration (AMD), diabetic retinopathy (DR), and glaucoma.
- cluster of differentiation 36
- post translational modification
- signaling pathway
- retina
- eye disease
1. Fundus Diseases and Pathological Changes
1.1. CD36 and Age-Related Macular Degeneration (AMD)
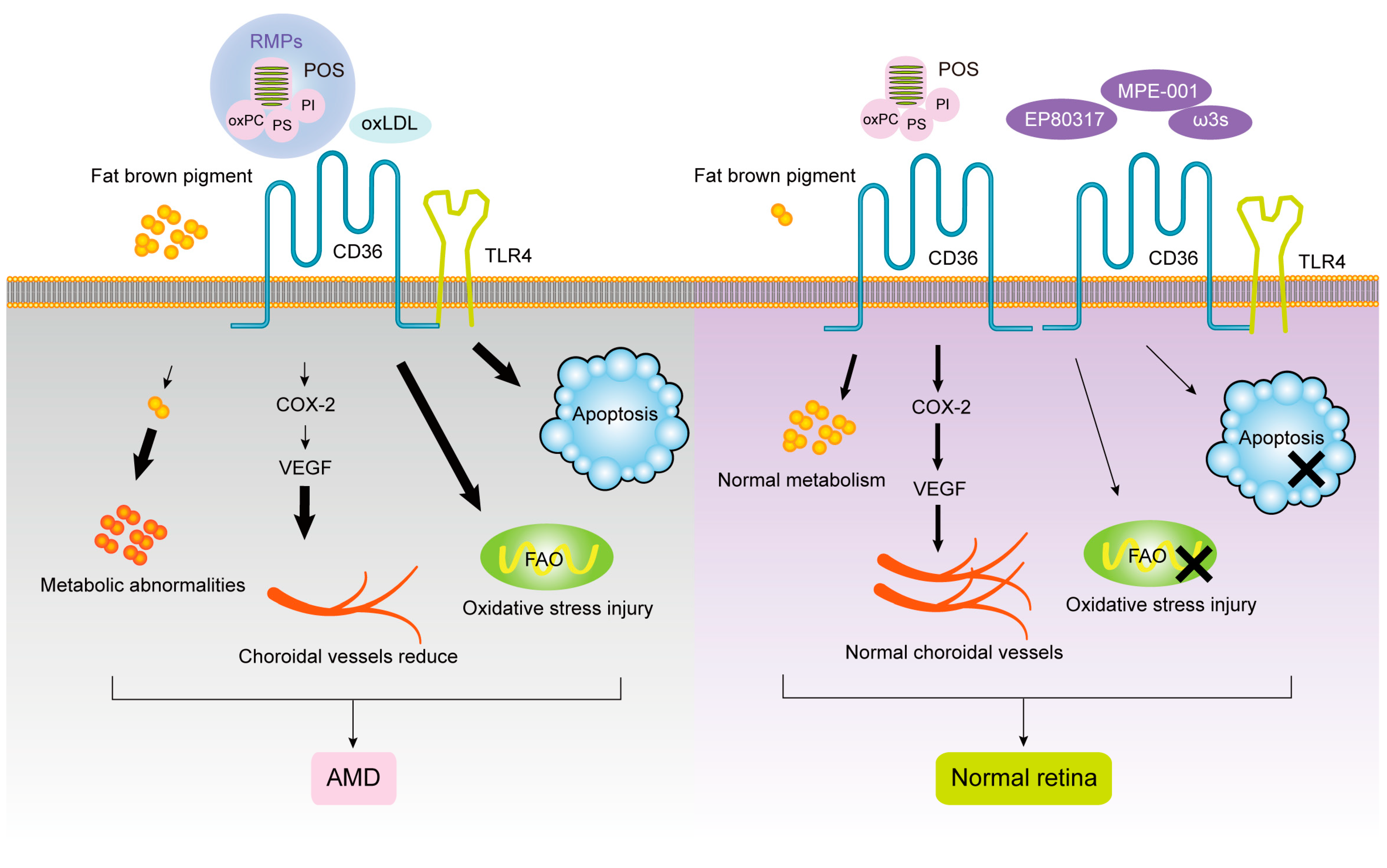
1.2. CD36 and Diabetic Retinopathy (DR)
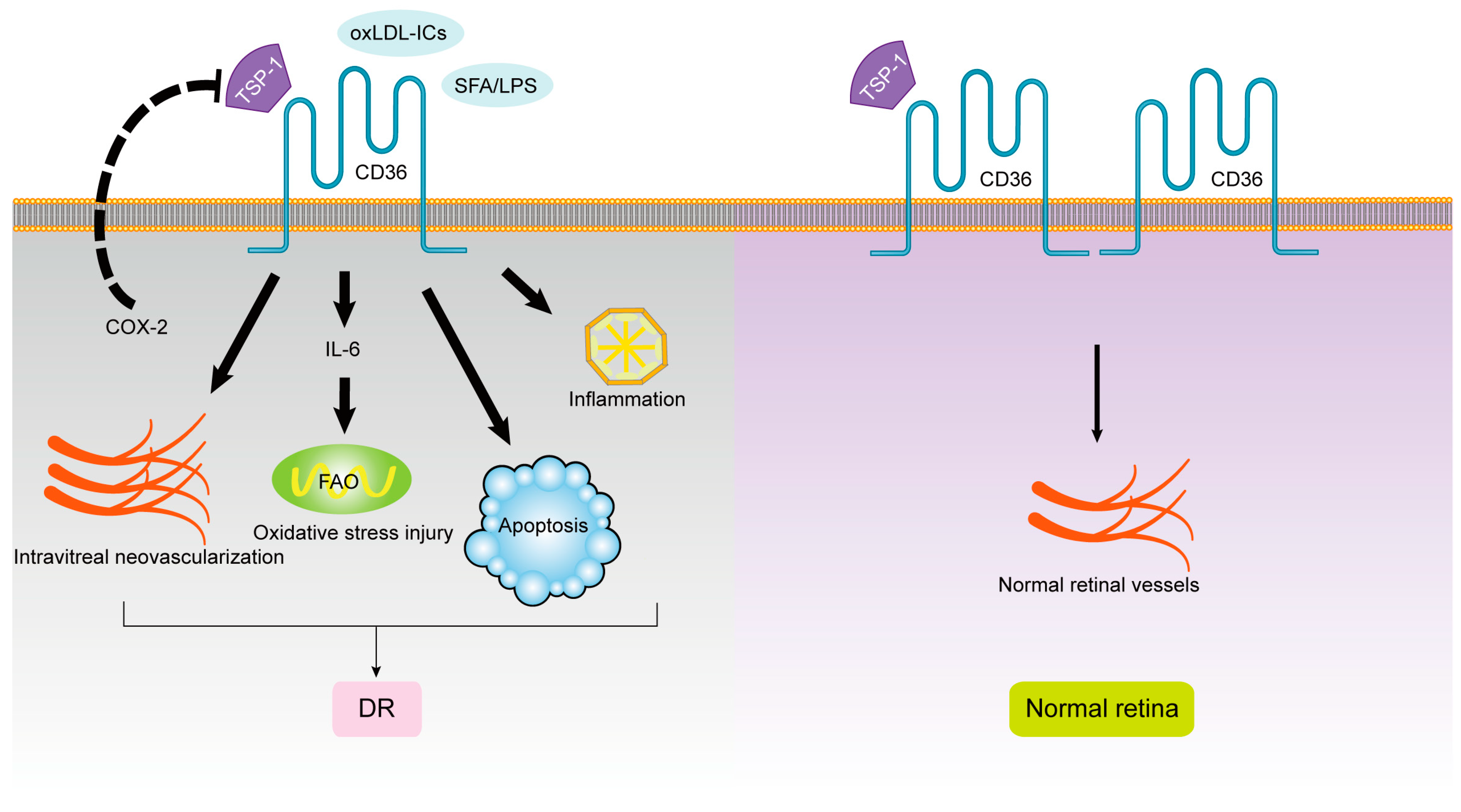
1.3. CD36 and Glaucoma
1.4. CD36 and Retinal Neovascularization
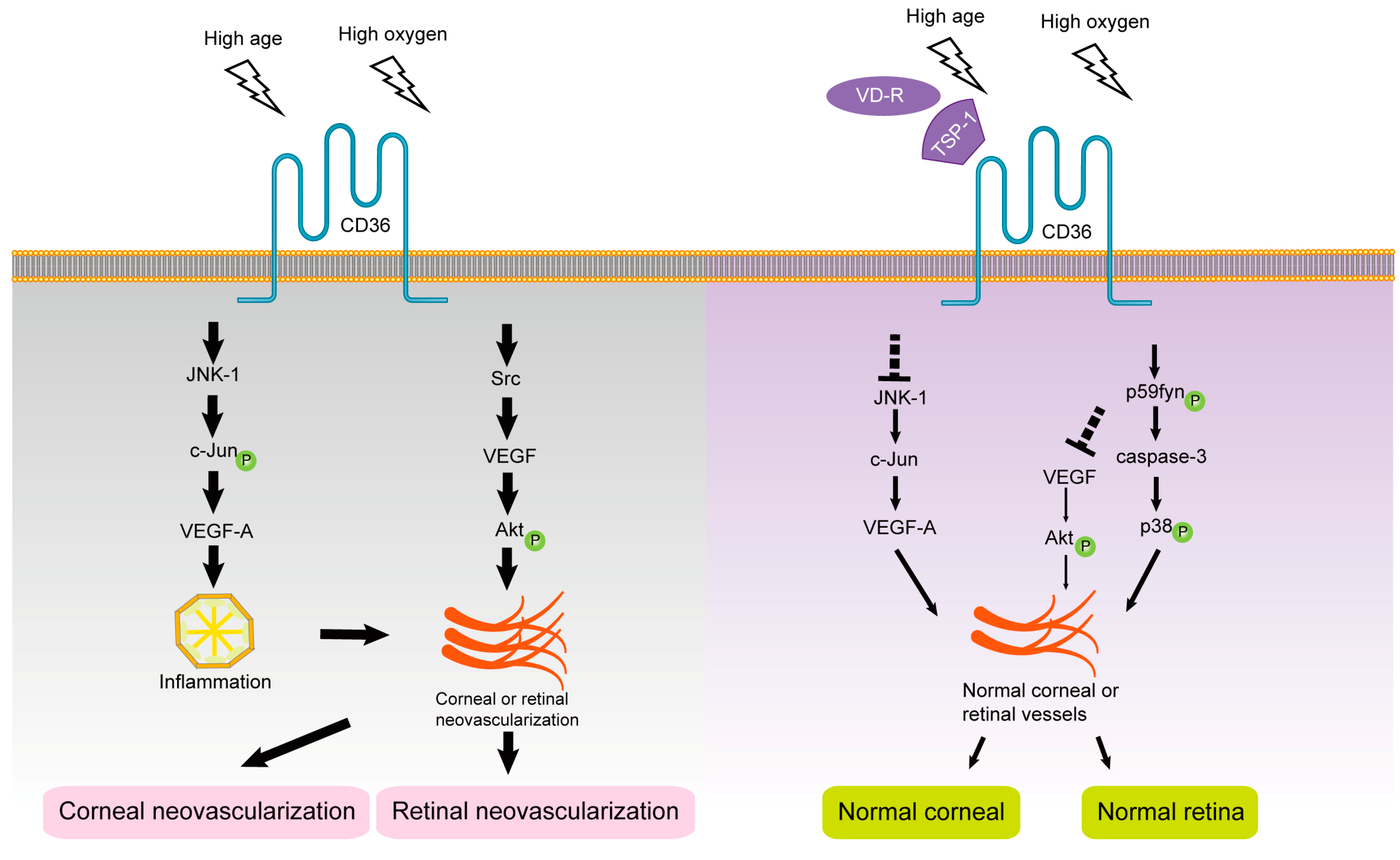
1.5. CD36 and Subretinal Inflammation
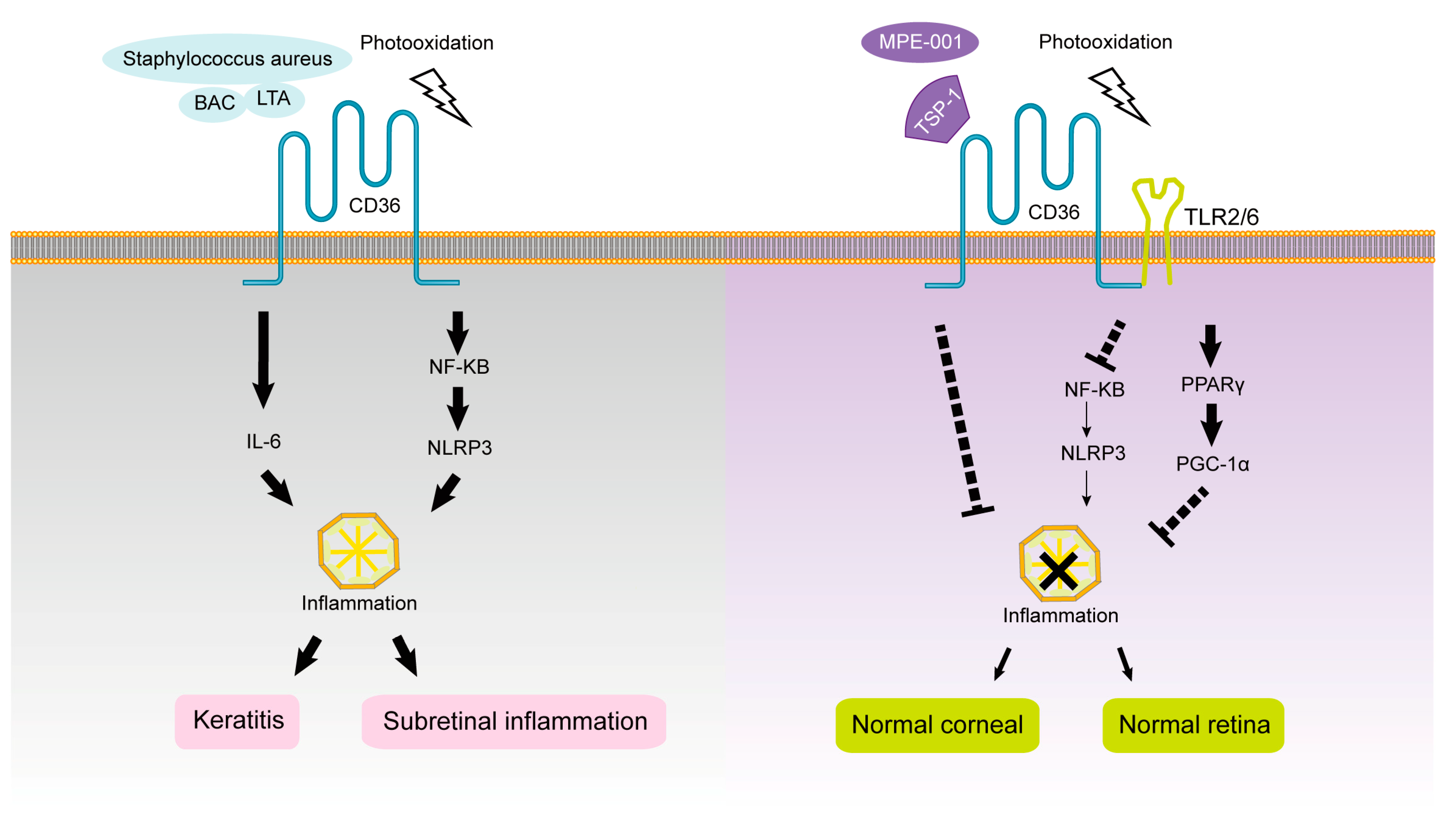
2. Ocular Surface Diseases and Pathological Changes
2.1. CD36 and Corneal Neovascularization (CNV)
2.2. CD36 and Keratitis
This entry is adapted from the peer-reviewed paper 10.3390/cells12010171
References
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805.
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917.
- Segawa, K.; Nagata, S. An Apoptotic ’Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650.
- Penberthy, K.K.; Lysiak, J.J.; Ravichandran, K.S. Rethinking Phagocytes: Clues from the Retina and Testes. Trends Cell Biol. 2018, 28, 317–327.
- Dorion, M.F.; Mulumba, M.; Kasai, S.; Itoh, K.; Lubell, W.D.; Ong, H. The CD36 Ligand-Promoted Autophagy Protects Retinal Pigment Epithelial Cells from Oxidative Stress. Oxidative Med. Cell. Longev. 2021, 2021, 6691402.
- Roggia, M.F.; Ueta, T. αvβ5 Integrin/FAK/PGC-1α Pathway Confers Protective Effects on Retinal Pigment Epithelium. PLoS ONE 2015, 10, e0134870.
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Veréb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984.
- Rieu, Q.; Bougoüin, A.; Zagar, Y.; Chatagnon, J.; Hamieh, A.; Enderlin, J.; Huby, T.; Nandrot, E.F. Pleiotropic Roles of Scavenger Receptors in Circadian Retinal Phagocytosis: A New Function for Lysosomal SR-B2/LIMP-2 at the RPE Cell Surface. Int. J. Mol. Sci. 2022, 23, 3445.
- Yang, C.; Shani, S.; Tahiri, H.; Ortiz, C.; Gu, M.; Lavoie, J.C.; Croteau, S.; Hardy, P. Extracellular microparticles exacerbate oxidative damage to retinal pigment epithelial cells. Exp. Cell Res. 2020, 390, 111957.
- Nandrot, E.F.; Kim, Y.; Brodie, S.E.; Huang, X.; Sheppard, D.; Finnemann, S.C. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking alphavbeta5 integrin. J. Exp. Med. 2004, 200, 1539–1545.
- Vollrath, D.; Feng, W.; Duncan, J.L.; Yasumura, D.; D’Cruz, P.M.; Chappelow, A.; Matthes, M.T.; Kay, M.A.; LaVail, M.M. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc. Natl. Acad. Sci. USA 2001, 98, 12584–12589.
- Ryeom, S.W.; Silverstein, R.L.; Scotto, A.; Sparrow, J.R. Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J. Biol. Chem. 1996, 271, 20536–20539.
- Ren, Y.; Silverstein, R.L.; Allen, J.; Savill, J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J. Exp. Med. 1995, 181, 1857–1862.
- Lin, H.; Clegg, D.O. Integrin alphavbeta5 participates in the binding of photoreceptor rod outer segments during phagocytosis by cultured human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1703–1712.
- Duncan, K.G.; Bailey, K.R.; Kane, J.P.; Schwartz, D.M. Human retinal pigment epithelial cells express scavenger receptors BI and BII. Biochem. Biophys. Res. Commun. 2002, 292, 1017–1022.
- Westenskow, P.D.; Moreno, S.K.; Krohne, T.U.; Kurihara, T.; Zhu, S.; Zhang, Z.N.; Zhao, T.; Xu, Y.; Ding, S.; Friedlander, M. Using flow cytometry to compare the dynamics of photoreceptor outer segment phagocytosis in iPS-derived RPE cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6282–6290.
- Finnemann, S.C.; Silverstein, R.L. Differential roles of CD36 and alphavbeta5 integrin in photoreceptor phagocytosis by the retinal pigment epithelium. J. Exp. Med. 2001, 194, 1289–1298.
- Chang, Y.; Finnemann, S.C. Tetraspanin CD81 is required for the alpha v beta5-integrin-dependent particle-binding step of RPE phagocytosis. J. Cell Sci. 2007, 120, 3053–3063.
- Sun, M.; Finnemann, S.C.; Febbraio, M.; Shan, L.; Annangudi, S.P.; Podrez, E.A.; Hoppe, G.; Darrow, R.; Organisciak, D.T.; Salomon, R.G.; et al. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J. Biol. Chem. 2006, 281, 4222–4230.
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Febbraio, M.; Hajjar, D.P.; et al. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 2002, 277, 38517–38523.
- Gordiyenko, N.; Campos, M.; Lee, J.W.; Fariss, R.N.; Sztein, J.; Rodriguez, I.R. RPE cells internalize low-density lipoprotein (LDL) and oxidized LDL (oxLDL) in large quantities in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2822–2829.
- Courtois, Y. The role of CD36 receptor in the phagocytosis of oxidized lipids and AMD. Aging 2010, 2, 888–889.
- Rigotti, A.; Acton, S.L.; Krieger, M. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J. Biol. Chem. 1995, 270, 16221–16224.
- Houssier, M.; Raoul, W.; Lavalette, S.; Keller, N.; Guillonneau, X.; Baragatti, B.; Jonet, L.; Jeanny, J.C.; Behar-Cohen, F.; Coceani, F.; et al. CD36 deficiency leads to choroidal involution via COX2 down-regulation in rodents. PLoS Med. 2008, 5, e39.
- Martini, C.; DeNichilo, M.; King, D.P.; Cockshell, M.P.; Ebert, B.; Dale, B.; Ebert, L.M.; Woods, A.; Bonder, C.S. CD36 promotes vasculogenic mimicry in melanoma by mediating adhesion to the extracellular matrix. BMC Cancer 2021, 21, 765.
- Kondo, N.; Honda, S.; Kuno, S.; Negi, A. Positive association of common variants in CD36 with neovascular age-related macular degeneration. Aging 2009, 1, 266–274.
- Honda, S.; Bessho, H.; Kondo, N.; Kusuhara, S.; Tsukahara, Y.; Negi, A. Positive association of CD36 gene variants with the visual outcome of photodynamic therapy in polypoidal choroidal vasculopathy. Mol. Vis. 2012, 18, 2796–2804.
- Yanagi, Y.; Foo, V.H.X.; Yoshida, A. Asian age-related macular degeneration: From basic science research perspective. Eye (Lond. Engl.) 2019, 33, 34–49.
- Bowers, C.Y. Growth hormone-releasing peptide (GHRP). Cell. Mol. Life Sci. 1998, 54, 1316–1329.
- Picard, E.; Houssier, M.; Bujold, K.; Sapieha, P.; Lubell, W.; Dorfman, A.; Racine, J.; Hardy, P.; Febbraio, M.; Lachapelle, P.; et al. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging 2010, 2, 981–989.
- Proulx, C.; Picard, É.; Boeglin, D.; Pohankova, P.; Chemtob, S.; Ong, H.; Lubell, W.D. Azapeptide analogues of the growth hormone releasing peptide 6 as cluster of differentiation 36 receptor ligands with reduced affinity for the growth hormone secretagogue receptor 1a. J. Med. Chem. 2012, 55, 6502–6511.
- Kindzelskii, A.L.; Elner, V.M.; Elner, S.G.; Yang, D.; Hughes, B.A.; Petty, H.R. Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J. Gen. Physiol. 2004, 124, 139–149.
- Cogan, D.G.; Toussaint, D.; Kuwabara, T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch. Ophthalmol. 1961, 66, 366–378.
- Adamis, A.P.; Berman, A.J. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin. Immunopathol. 2008, 30, 65–84.
- Adamiec-Mroczek, J.; Oficjalska-Młyńczak, J.; Misiuk-Hojło, M. Roles of endothelin-1 and selected proinflammatory cytokines in the pathogenesis of proliferative diabetic retinopathy: Analysis of vitreous samples. Cytokine 2010, 49, 269–274.
- Sennlaub, F.; Valamanesh, F.; Vazquez-Tello, A.; El-Asrar, A.M.; Checchin, D.; Brault, S.; Gobeil, F.; Beauchamp, M.H.; Mwaikambo, B.; Courtois, Y.; et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation 2003, 108, 198–204.
- Truman, J.P.; Al Gadban, M.M.; Smith, K.J.; Jenkins, R.W.; Mayroo, N.; Virella, G.; Lopes-Virella, M.F.; Bielawska, A.; Hannun, Y.A.; Hammad, S.M. Differential regulation of acid sphingomyelinase in macrophages stimulated with oxidized low-density lipoprotein (LDL) and oxidized LDL immune complexes: Role in phagocytosis and cytokine release. Immunology 2012, 136, 30–45.
- Lopes-Virella, M.F.; Baker, N.L.; Hunt, K.J.; Lyons, T.J.; Jenkins, A.J.; Virella, G. High concentrations of AGE-LDL and oxidized LDL in circulating immune complexes are associated with progression of retinopathy in type 1 diabetes. Diabetes Care 2012, 35, 1333–1340.
- Fredrikson, G.N.; Anand, D.V.; Hopkins, D.; Corder, R.; Alm, R.; Bengtsson, E.; Shah, P.K.; Lahiri, A.; Nilsson, J. Associations between autoantibodies against apolipoprotein B-100 peptides and vascular complications in patients with type 2 diabetes. Diabetologia 2009, 52, 1426–1433.
- Fu, D.; Yu, J.Y.; Wu, M.; Du, M.; Chen, Y.; Abdelsamie, S.A.; Li, Y.; Chen, J.; Boulton, M.E.; Ma, J.X.; et al. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J. Lipid Res. 2014, 55, 860–869.
- Abdelsamie, S.A.; Li, Y.; Huang, Y.; Lee, M.H.; Klein, R.L.; Virella, G.; Lopes-Virella, M.F. Oxidized LDL immune complexes stimulate collagen IV production in mesangial cells via Fc gamma receptors I and III. Clin. Immunol. 2011, 139, 258–266.
- Ralston, J.C.; Metherel, A.H.; Stark, K.D.; Mutch, D.M. SCD1 mediates the influence of exogenous saturated and monounsaturated fatty acids in adipocytes: Effects on cellular stress, inflammatory markers and fatty acid elongation. J. Nutr. Biochem. 2016, 27, 241–248.
- Sasaki, M.; Kawasaki, R.; Rogers, S.; Man, R.E.; Itakura, K.; Xie, J.; Flood, V.; Tsubota, K.; Lamoureux, E.; Wang, J.J. The Associations of Dietary Intake of Polyunsaturated Fatty Acids With Diabetic Retinopathy in Well-Controlled Diabetes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7473–7479.
- Xu, C.; Chakravarty, K.; Kong, X.; Tuy, T.T.; Arinze, I.J.; Bone, F.; Massillon, D. Several transcription factors are recruited to the glucose-6-phosphatase gene promoter in response to palmitate in rat hepatocytes and H4IIE cells. J. Nutr. 2007, 137, 554–559.
- Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Interaction of palmitate and LPS regulates cytokine expression and apoptosis through sphingolipids in human retinal microvascular endothelial cells. Exp. Eye Res. 2019, 178, 61–71.
- Baranova, I.N.; Kurlander, R.; Bocharov, A.V.; Vishnyakova, T.G.; Chen, Z.; Remaley, A.T.; Csako, G.; Patterson, A.P.; Eggerman, T.L. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J. Immunol. 2008, 181, 7147–7156.
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674.
- Wilkinson, K.; Boyd, J.D.; Glicksman, M.; Moore, K.J.; El Khoury, J. A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J. Biol. Chem. 2011, 286, 34914–34922.
- Howlett, D.R.; Bate, S.T.; Collier, S.; Lawman, A.; Chapman, T.; Ashmeade, T.; Marshall, I.; Anderson, P.J.; Philpott, K.L.; Richardson, J.C.; et al. Characterisation of amyloid-induced inflammatory responses in the rat retina. Exp. Brain Res. 2011, 214, 185–197.
- Simons, E.S.; Smith, M.A.; Dengler-Crish, C.M.; Crish, S.D. Retinal ganglion cell loss and gliosis in the retinofugal projection following intravitreal exposure to amyloid-beta. Neurobiol. Dis. 2021, 147, 105146.
- Lawler, J. The functions of thrombospondin-1 and-2. Curr. Opin. Cell Biol. 2000, 12, 634–640.
- Chen, H.; Herndon, M.E.; Lawler, J. The cell biology of thrombospondin-1. Matrix Biol. 2000, 19, 597–614.
- Jiménez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48.
- Tian, R.; Deng, A.; Pang, X.; Chen, Y.; Gao, Y.; Liu, H.; Hu, Z. VR-10 polypeptide interacts with CD36 to induce cell apoptosis and autophagy in choroid-retinal endothelial cells: Identification of VR-10 as putative novel therapeutic agent for choroid neovascularization (CNV) treatment. Peptides 2022, 157, 170868.
- Upalakalin, J.N.; Hemo, I.; Dehio, C.; Keshet, E.; Benjamin, L.E. Survival mechanisms of VEGF and PlGF during microvascular remodeling. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 181–187.
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832.
- Sun, J.; Hopkins, B.D.; Tsujikawa, K.; Perruzzi, C.; Adini, I.; Swerlick, R.; Bornstein, P.; Lawler, J.; Benjamin, L.E. Thrombospondin-1 modulates VEGF-A-mediated Akt signaling and capillary survival in the developing retina. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1344–H1351.
- Dong, Y.; Cai, X.; Wu, Y.; Liu, Y.; Deng, L.; Chen, H. Insights from Genetic Model Systems of Retinal Degeneration: Role of Epsins in Retinal Angiogenesis and VEGFR2 Signaling. J. Nat. Sci. 2017, 3.
- Whitcup, S.M.; Nussenblatt, R.B.; Lightman, S.L.; Hollander, D.A. Inflammation in retinal disease. Int. J. Inflamm. 2013, 2013, 724648.
- Abe, T.; Shimamura, M.; Jackman, K.; Kurinami, H.; Anrather, J.; Zhou, P.; Iadecola, C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke 2010, 41, 898–904.
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242.
- Mellal, K.; Omri, S.; Mulumba, M.; Tahiri, H.; Fortin, C.; Dorion, M.F.; Pham, H.; Garcia Ramos, Y.; Zhang, J.; Pundir, S.; et al. Immunometabolic modulation of retinal inflammation by CD36 ligand. Sci. Rep. 2019, 9, 12903.
- Lavalette, S.; Conart, J.B.; Touhami, S.; Roubeix, C.; Houssier, M.; Augustin, S.; Raoul, W.; Combadière, C.; Febbraio, M.; Ong, H.; et al. CD36 Deficiency Inhibits Retinal Inflammation and Retinal Degeneration in Cx3cr1 Knockout Mice. Front. Immunol. 2019, 10, 3032.
- Nicholas, M.P.; Mysore, N. Corneal neovascularization. Exp. Eye Res. 2021, 202, 108363.
- Ren, S.W.; Qi, X.; Jia, C.K.; Wang, Y.Q. Serum amyloid A and pairing formyl peptide receptor 2 are expressed in corneas and involved in inflammation-mediated neovascularization. Int. J. Ophthalmol. 2014, 7, 187–193.
- Jia, C.; Zhu, W.; Ren, S.; Xi, H.; Li, S.; Wang, Y. Comparison of genome-wide gene expression in suture- and alkali burn-induced murine corneal neovascularization. Mol. Vis. 2011, 17, 2386–2399.
- Mwaikambo, B.R.; Yang, C.; Ong, H.; Chemtob, S.; Hardy, P. Emerging roles for the CD36 scavenger receptor as a potential therapeutic target for corneal neovascularization. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 255–272.
- Mwaikambo, B.R.; Sennlaub, F.; Ong, H.; Chemtob, S.; Hardy, P. Genetic ablation of CD36 induces age-related corneal neovascularization. Cornea 2008, 27, 1037–1041.
- Klocke, J.; Barcia, R.N.; Heimer, S.; Cario, E.; Zieske, J.; Gilmore, M.S.; Ksander, B.R.; Gregory, M.S. Spontaneous bacterial keratitis in CD36 knockout mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 256–263.
- Ricciuto, J.; Heimer, S.R.; Gilmore, M.S.; Argüeso, P. Cell surface O-glycans limit Staphylococcus aureus adherence to corneal epithelial cells. Infect. Immun. 2008, 76, 5215–5220.
- Mwaikambo, B.R.; Sennlaub, F.; Ong, H.; Chemtob, S.; Hardy, P. Activation of CD36 inhibits and induces regression of inflammatory corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4356–4364.
- Yoon, H.; Choi, S.I.; Kim, E.K. Uptake of cell debris and enhanced expression of inflammatory factors in response to dead cells in corneal fibroblast cells. Exp. Eye Res. 2020, 194, 108017.
- Soriano-Romaní, L.; García-Posadas, L.; López-García, A.; Paraoan, L.; Diebold, Y. Thrombospondin-1 induces differential response in human corneal and conjunctival epithelial cells lines under in vitro inflammatory and apoptotic conditions. Exp. Eye Res. 2015, 134, 1–14.