Reactive oxygen species (ROS) have been described to induce a broad range of redox-dependent signaling reactions in physiological conditions. Nevertheless, an excessive accumulation of ROS leads to oxidative stress, which was traditionally considered as detrimental for cells and organisms, due to the oxidative damage they cause to biomolecules. During ageing, elevated ROS levels result in the accumulation of damaged proteins, which may exhibit altered enzymatic function or physical properties (e.g., aggregation propensity). Emerging evidence also highlights the relationship between oxidative stress and age-related pathologies, such as protein misfolding-based neurodegenerative diseases (e.g., Parkinson’s (PD), Alzheimer’s (AD) and Huntington’s (HD) diseases).
1. Introduction
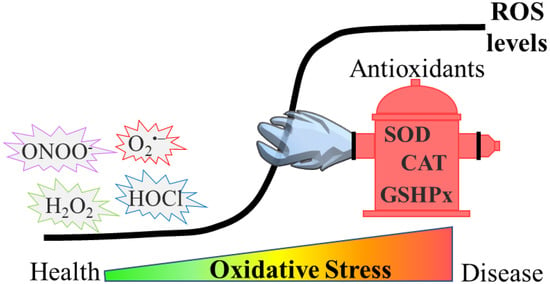
Oxidative stress can be defined as an imbalance between the generation of reactive oxygen species (ROS) and their quenching by antioxidants in a specific cell or tissue (
Figure 1). One of the most studied ROS is hydrogen peroxide (H
2O
2), but many other, more reactive species, such as superoxide anion radicals (O
2•−), hydroxyl radicals (
●OH), singlet oxygen (
1O
2), nitrogen dioxide (NO
2●−), hypochlorous acid (HOCl) and peroxynitrite (ONOO
−) are generated by various enzymatic and non-enzymatic processes in biological systems [
1].
Figure 1. Development of oxidative stress. In physiological conditions, ROS remain at low levels by the effect of antioxidants such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSHPx). However, during oxidative stress, ROS levels rise, due to either higher production or impaired antioxidant defense (or both), and this results in the accumulation of damaged biomolecules. Such events are detrimental and either drive or exacerbate diseases such as neurodegenerative disorders.
The excessive amount of ROS triggers the so-called redox-dependent signaling in physiological conditions. However, when it surpasses a threshold, ROS become detrimental. Apart from aberrant redox signaling, ROS can damage a myriad of cellular components, such as proteins, DNA and lipids. As several amino acid residues are amenable to oxidation (depending on the ROS involved), oxidative damage leads to a variety of oxidative modifications onto proteins. These modifications could alter cellular function by regulating stability, activity, subcellular localization or the protein–protein interaction of oxidized proteins [
2]. A wide range of cellular mechanisms are activated to eliminate ROS as well as the ROS-mediated oxidized biomolecules (such as the newly discovered protection of promoters from oxidative damage though the action of the nuclear mitotic apparatus protein, NuMA [
3]). Nonetheless, upon persistent activation these mechanisms may either aggravate the cellular damage or become impaired, eventually leading to a vicious cycle that promotes the progression of several pathologies.
Oxidative stress is implicated in a wide variety of age-related diseases ranging from diabetes and cardiovascular disease to cancer and neurodegenerative diseases [
4,
5,
6]. A common characteristic of most neurodegenerative diseases is the formation of toxic protein aggregates that affect many functions, eventually leading to neuronal loss. Interestingly, several of these proteins can become oxidized. While this may not come as a surprise since protein oxidative damage is a non-discriminative process, studies on α- synuclein [
7], prion protein [
8] and TDP-43 [
9] have shown that their oxidation is in fact required for their initial nucleation to take place. This may indicate that the oxidation of aggregation-prone proteins may constitute another common characteristic among neurological disorders. The role of oxidative stress in these diseases is further evident by the fact that oxidative damage has been consistently detected in the post-mortem brains of patients with Alzheimer’s (AD), Parkinson’s (PD) and Huntington’s (HD) diseases [
10]. While it is currently unknown whether oxidative stress is a driver or a consequence that further exacerbates them, these findings point to the dependency of disease progression on oxidative stress.
2. Oxidative Stress in Ageing and Age-Related Neurodegenerative Diseases
In numerous diseases, the amount of ROS is increased and poses a threat to cells and organisms. Oxidative damage has been suggested as both an effect and a mediator of ageing and a range of age-related neurodegenerative pathological conditions [
93].
Nutraceuticals treatment of model organisms has remarkably contributed to elucidating the physiological mechanisms associated with biological ageing. For example, data from the nematode
Caenorhabditis elegans conclude that the effects of polyphenolic phytochemical phlorizin (phloridzin) diminish the intracellular ROS levels (measured by H
2DCF-DA). This decrease in ROS activates a DAF-16-induced stress response and autophagy, subsequently leading to lifespan extension. Moreover, supplementation with phloridzin reverses the inactivation of dopaminergic neurons observed in worm models of PD [
94]. Other compounds, such as the natural product cryptotanshinone [
95] or the glucagon-like peptide-1 receptor (GLP-1R) agonist Exendin-4 [
96], have been tested in
Caenorhabditis elegans models of AD, concluding that those drugs can decrease oxidative stress and protein aggregation and improve the disease phenotype. In the following paragraphs we will provide examples of how oxidative stress is implicated in disease manifestations in three age-related neurodegenerative disorders, namely PD, AD and HD.
PD is a progressive age-related neurodegenerative condition associated with a loss of dopaminergic neurons in the substantia nigra pars compacta of the brain. The symptomatology gradually increases upon onset of the disease and is mainly related to motor problems. Although the specific molecular mechanisms underlying the loss of dopaminergic neurons is not fully understood, oxidative stress is a well-established trigger of dopaminergic neurotoxicity [
97].
In this line, Soto-Rojas and co-workers determined ROS (through detection of the fluorescence signal of 2,7-dichloro dihydrofluorescein diacetate [DCFH-DA]) and lipid peroxidation (by evaluating the formation of lipid-soluble fluorescent compounds) in the brain of rats with α-synucleinopathy. They found that these two parameters of oxidative stress, as well as mitochondrial complex I dysfunction, positively correlate with neurodegeneration in different brain areas with α-synucleinopathy, such as the substantia nigra pars compacta, the striatum, the hippocampus and the olfactory bulb. Based on in silico studies, they suggested the involvement of peroxisome proliferator-activated receptors (PPAR) alpha (PPAR-α) and gamma (PPAR-γ) in this process [
98], since PPARs activation induces fatty acid oxidation (which, in turn, generates ROS) [
99]. However, this mechanism needs to be further explored. Two additional recent studies further highlighted the interplay between ROS and PD. Bardien and co-workers sought to understand the pathophysiological mechanisms associated with familiar autosomal dominant PD. More specifically, they studied the p.G849D variant in the neurexin 2α (
NRXN2) gene, which has been described to co-segregate with PD. Using a cellular model carrying the neurexin 2α p.G849D variant, the authors found elevation of H
2O
2 levels (using a commercially available assay), accompanied by neuronal death [
100]. Saiki and co-workers wished to shed light on the mechanism that promotes lysosomal retrograde transport, a regulator of autophagy, in PD. The authors initially demonstrated that JIP4 is upstream of the TRPML1-ALG2 pathway, which is known to promote retrograde transport. Subsequently, it was shown that this pathway can be activated in a ROS-dependent manner, since treatment with H
2O
2 induces the phosphorylation of JIP4. These results highlight that oxidative stress could, in part, regulate autophagy in the context of PD via the JIP4-TRPML1-ALG2 pathway [
101].
AD is a chronic, progressive neurological disorder that is associated with intraneuronal filamentous inclusions, called neurofibrillary tangles, mostly composed by tau protein aggregates, and extracellular senile amyloid plaques, which are predominantly due to the aggregation of misfolded Aβ peptide. Among neurodegenerative diseases, AD is the most common cause of dementia [
102]. Similar to PD, alterations in oxidative stress and mitochondrial dynamics and function are known to play a role in the development of the disease [
103]. Studies using in vitro models of AD have shown that treatment with the ROS scavenger N-acetyl-L-cysteine (NAC) reduces Aβ deposition through the activation of the PI3K/Akt/GLUT1 pathway, while it ameliorates the impaired glucose uptake phenotype. In this case, the authors measured intracellular ROS levels by flow cytometry [
104].
HD is an autosomal dominant neurodegenerative disorder caused by CAG triplet expansions in the
Huntingtin gene, encoding an elongated poly-glutamine stretch in the Huntingtin protein. Patients with HD exhibit motor, psychiatric and cognitive deterioration and oxidative stress, although it is not clear whether it is the cause or the consequence of disease progression [
105]. Ellederová and co-workers found higher oxidative stress in fibroblasts from a minipig model of HD, compared with wild-type minipig fibroblasts. The authors measured ROS (by using CellROX deep red reagent), lipid peroxidation (with Image-iT) and membrane permeability (with Calcein-AM). These elevated levels of oxidative stress correlated with the overexpression of mitochondrial superoxide dismutase 2 (SOD2) and the NEIL3 gene (encoding DNA glycosylase). SOD2 is expressed in the mitochondria, where it controls the cell cycle as a response to oxidative stress, whereas glycosylase enzyme repairs in the replication-associated repair of oxidized DNA. Taken together, these data highlight the role of oxidative stress and DNA damage in HD [
106].
Southwell and co-workers overexpressed progerin, a protein that causes premature aging, in murine neurons with HD, and observed ageing-related phenotypes. By treating neurons with H
2O
2 and quantifying ROS (with CM-H
2DCFDA), the authors concluded that biological ageing increases the sensitivity of neurons with HD to exogenous oxidative stress [
107,
108]. This sensitivity may highlight the interplay between ROS and protein aggregation, as shown for other neurodegenerative diseases.
In the same line, Oliveira and co-workers used MitoParaquat (MitoPQ) to enhance mitochondrial superoxide production. They observed features of PD in MitoPQ-treated zebrafish and an induction of mutant huntingtin aggregation without increasing cell death in a human cell model of HD, upon treatment with MitoPQ [
109]. The forkhead box O1 (FOXO1) transcription factor has been shown to be involved in the development of AD and HD. Importantly, upon increased ROS, FOXO1 is activated through the AMPK pathway to promote autophagy, which is a well-established mechanism for the clearance of abnormal proteins and organelles [
110].
This entry is adapted from the peer-reviewed paper 10.3390/antiox12010131