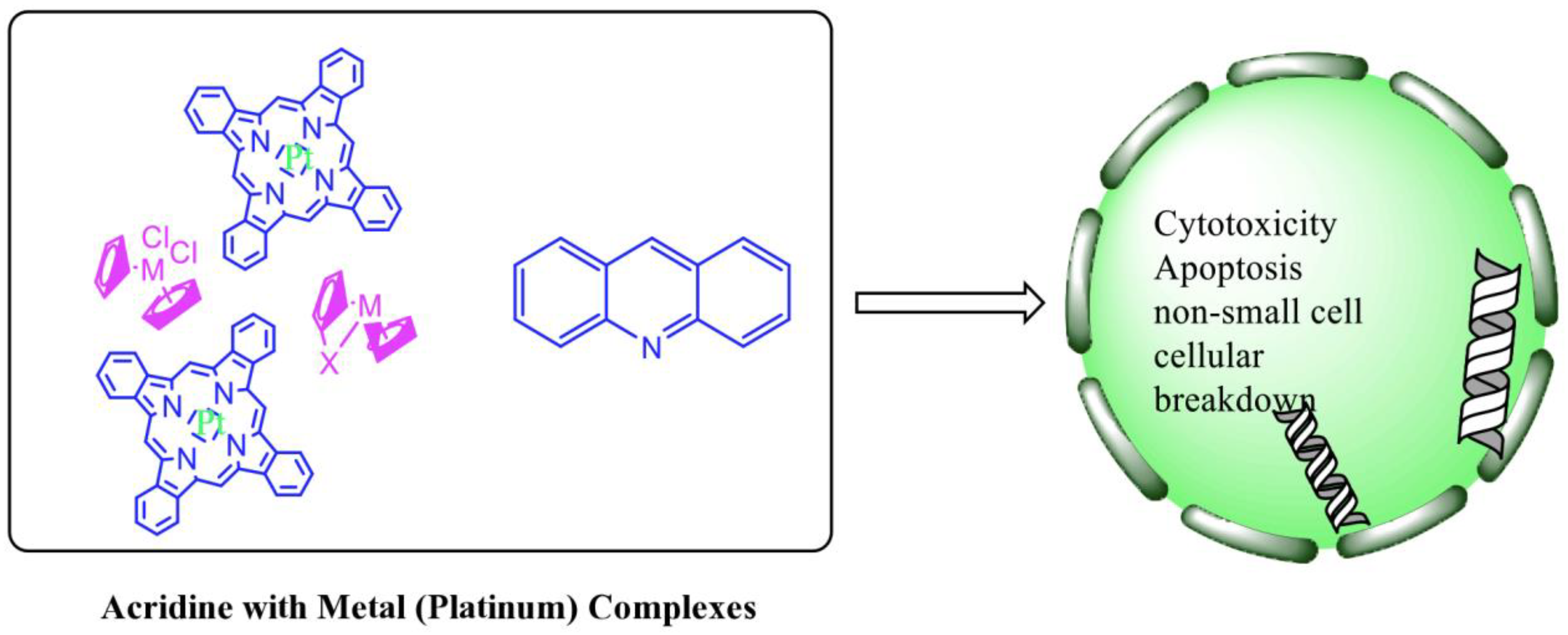
Acridine derivatives are a class of compounds that are being extensively researched as potential anti-cancer drugs. Acridines are well-known for their high cytotoxic activity; however, their clinical application is restricted or even excluded as a result of side effects. The photocytotoxicity of propyl acridine acts against leukaemia cell lines, with C1748 being a promising anti-tumour drug against UDP-UGT’s. CK0403 is reported in breast cancer treatment and is more potent than CK0402 against estrogen receptor-negative HER2. Acridine platinum (Pt) complexes have shown specificity on the evaluated DNA sequences; 9-anilinoacridine core, which intercalates DNA, and a methyl triazene DNA-methylating moiety were also studied. Acridine thiourea gold and acridinone derivatives act against cell lines such as MDA-MB-231, SK-BR-3, and MCF-7. Benzimidazole acridine compounds demonstrated cytotoxic activity against Dual Topo and PARP-1. Quinacrine, thiazacridine, and azacridine are reported as anti-cancer agents, which have been reported in the previous decade and were addressed in this research.
- acridine
- anti-cancer
- cancer cell lines
- DNA- intercalation
- topoisomerase
- in vitro assay
1. Introduction
2. Acridine as an Anti-Tumour Agent
2.1. Acridine
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Propyl-AcrDTU | 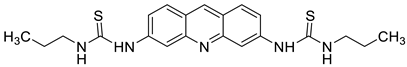 |
Leukaemia L1210 cells | [1] |
| Tetrandrine-based receptors | 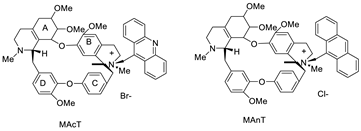 |
Anti-proliferative studies | [2] |
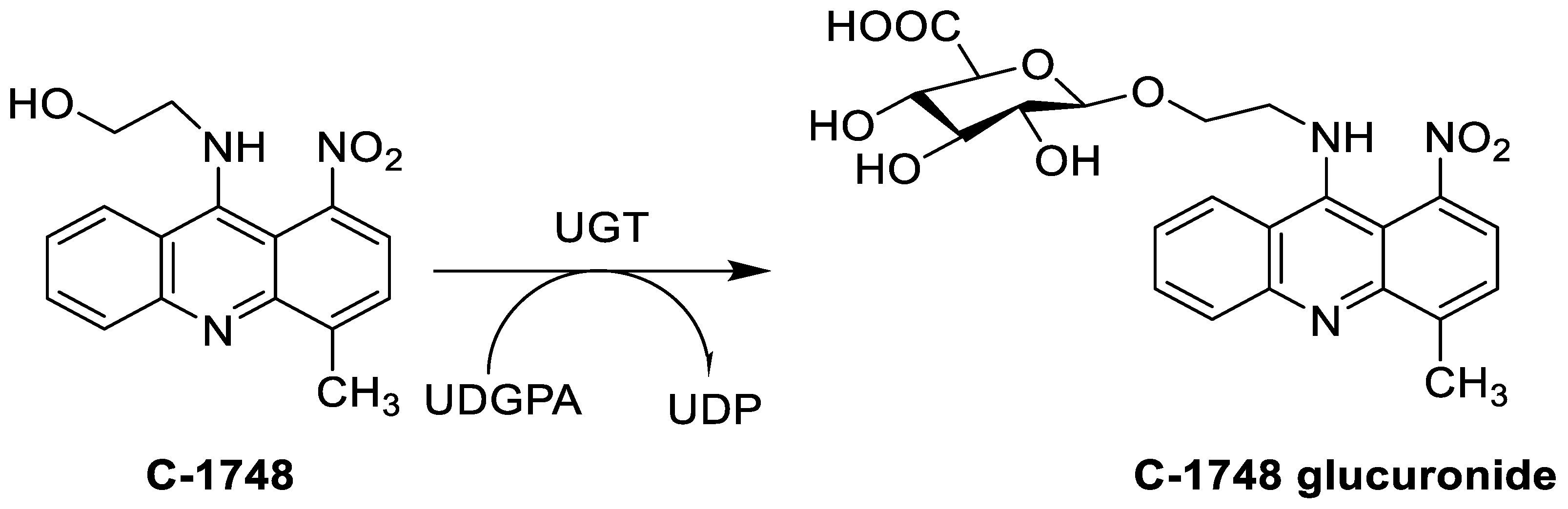
| 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C1748) | 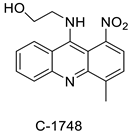 |
Phase II enzymes—UDP-glucuronosyltransferases (UGTs) | [3] |
| - |  |
Human lung adenocarcinoma cells | [4] |
| Acridine chalcone 1C ((2 E)-3-(acridin-9-yl)-1-(2,6-dimethoxyphenyl)prop-2-en-1-one) | 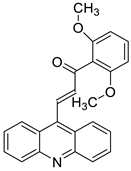 |
Human colorectal HCT116 cells | [5] |
| Novel spiro-acridine (E)-50-oxo-10-((3,4,5-trimethoxybenzylidene)amino)-10,50-dihydro-10H-spiro[acridine-9,20-pyrrole]40-carbonitrile (AMTAC-17) | 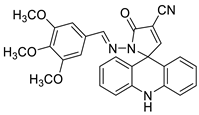 |
Anti-tumour activity of AMTAC-17 | [6] |
| 2-((6-Chloro-2-methoxy-acridin-9-yl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]-thiophene-3-carbonitrile (ACS03) | 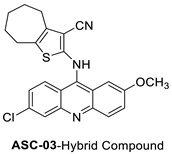 |
Anti-tumour activity | [7] |
| Dimethyl 2-[(acridin-9-yl) methylidene]-malonate (LPSF/IP-81) | 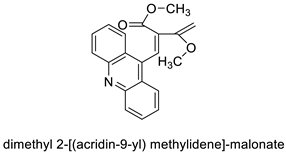 |
Invasive ductal carcinoma of human breast. | [8] |
| Bis (acridine-9-carboxylate)-nitro-europium (III) dihydrate | 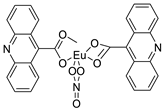 |
Anti-angiogenic and apoptotic activity against an animal model of carcinogenesis | [9] |
| 9-amino-1-nitroacridine | 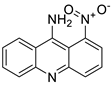 |
Pancreatic cancer | [10] |
| 9-amino acridine derivatives | 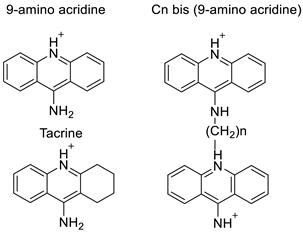 |
On alpha-adrenergic receptors | [11] |
| AT11-L0 | 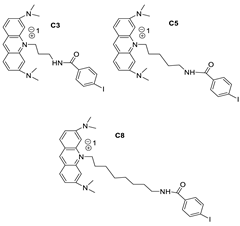 |
Anti-proliferative activity | [12] |
| Novel acridine-based N-acyl-homoserine lactone (AHL) analogs |  |
Human oral squamous carcinoma | [13] |
| New series of acridine hydroxamic acid derivatives |  |
Topo II inhibition activity | [14] |
| 9-chloro-2-(3-(dimethylamino) propyl) pyrrolo [2,3,4-kl] acridin-1(2H)-one (LS-1-10) | 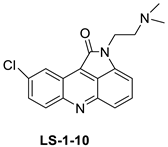 |
Colon cancer cells | [15] |
| Two iodinated acridine derivatives | 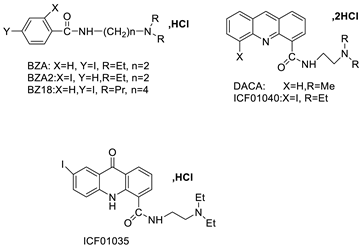 |
Targeted radionuclide therapy | [16] |
| [125I] ICF01035 and [125I] ICF01040 |  |
Melanoma-targeted in mice bearing B16F0 | [17] |
| 9-phenyl acridine (ACPH) | 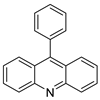 |
Anti-cancer agent | [18] |
| Tetra hydro acridine derivatives with iodobenzoic moiety | 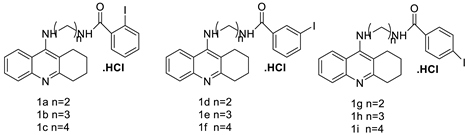 |
Human lung adenocarcinoma, human colorectal adenocarcinoma. | [19] |
| A series of 9-(2-(1-arylethylidene) hydrazinyl) acridine | 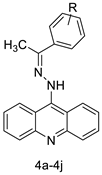 |
Anti-cancer activity | [20] |
| 9-phenylacridine (ACPH) | 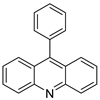 |
Antitumour activity | [21] |
| A class of acridine derivatives |  |
In vivo MM tumour growth | [22] |
| N0-(2-chloro-6-methoxy-acridin-9-yl)-2-cyano-3-(4-dimethylaminophenyl)-acrilohidrazida (ACS-AZ10). ACS-AZ10 | 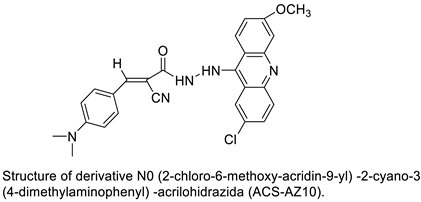 |
Anti-tumour activity | [23] |
| Acridine yellow G | 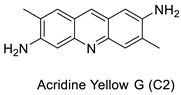 |
Inhibits both EGFR and PKCs | [24] |
| Unsymmetrical bisacridine derivatives (UAs) | 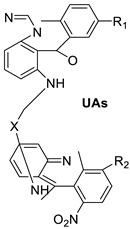 |
Cytotoxicity screening against several tumour cell lines | [25] |
| 9-aryl-hexahydro-acridine-1,8-diones | 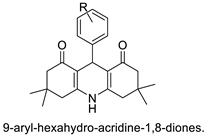 |
Anti-cancer activity against HepG2 and MCF-7 cell lines | [26] |
| Series of acridine-based catalytic inhibitors | 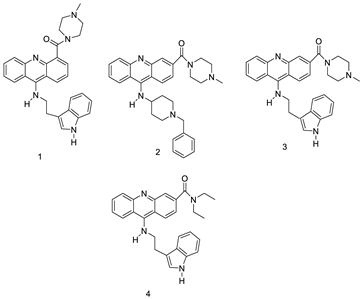 |
Pancreatic cancer and hTopoII catalytic inhibitors | [27] |
| Four hybrid acridine-HSP90 |  |
Telomerase inhibitor BRACO-19 | [28] |
| Three new diphenyl substituted spiro triazolidine- and thiazolidinone-acridines | 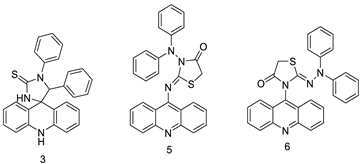 |
Interaction with calf thymus DNA | [29] |
| Amino acid appended acridines |  |
Anti-proliferative activity, arrested cells in G0/G1 phase of the cell cycle |
[30] |
| A novel series of tri-substituted acridines | 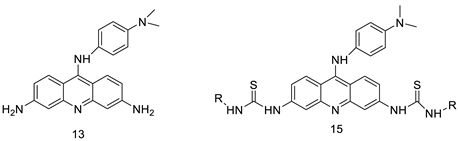 |
Mimicking the effects of BRACO19 | [31] |
| DNA–polymer hybrids |  |
DNA–polymerisation | [32] |
| A novel DNA-cleaving agent CuGGHK-Acr | 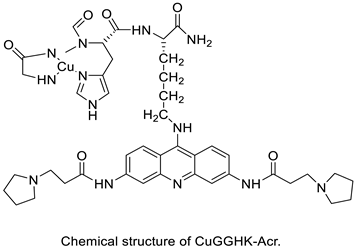 |
Targets G4 telomeric DNA | [33] |
| A series of 9-benzyl acridine derivatives | 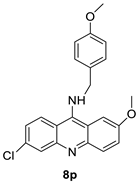 |
Anti-proliferative inhibitors | [34] |
2.2. 9-Amino Acridine
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Acridine-based catalytic inhibitors | 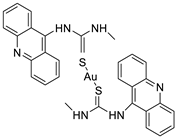 |
Anti-proliferative activity | [35] |
| CK0403 | 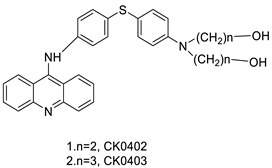 |
Treatment of breast cancer | [36] |
| 9-amino acridines | 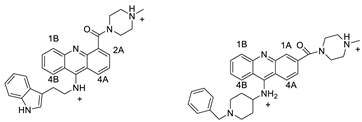 |
Anti-cancer activity | [37] |
| 9-aminoacridine and artemisinin–acridine | 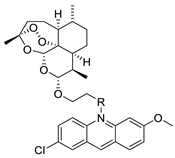 |
Anti-malarial activity, anti-cancer activity | [38] |
| 9-aminoacridine (9AA) | 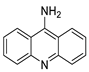 |
Anti-leukaemic cells | [39] |
| 9-aminoacridine carboxamide | 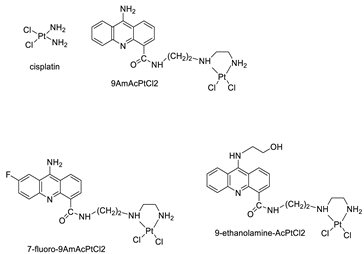 |
DNA-targeted intercalator | [40] |
| 9-aminoacridine derivatives | 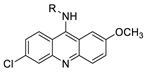 |
Anti-proliferative activity | [41] |
| Four acridine Pt complexes | 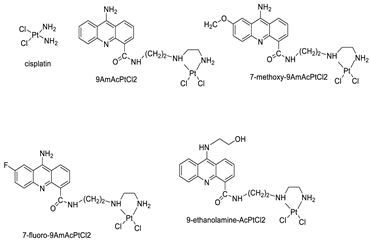 |
Detailed DNA sequence specificity | [42] |
| Small library of substituted 9-aminoacridine derivatives |  |
Ability to inhibit proliferation and induce cellular death in SCLC | [43] |
2.3. 9-Anilino Acridines
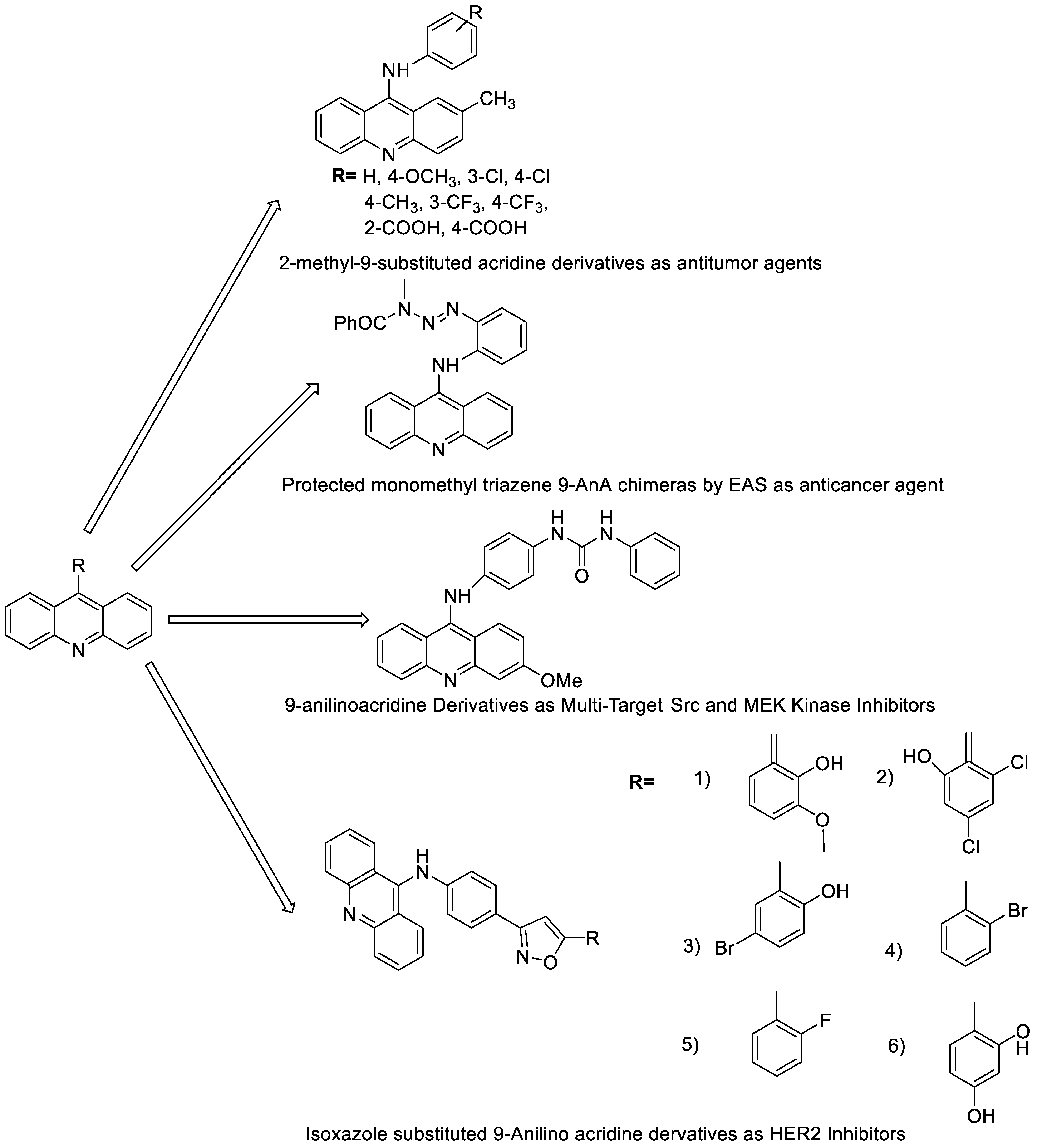
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| 2-methyl-9 substituted (AS 0–8) acridines | 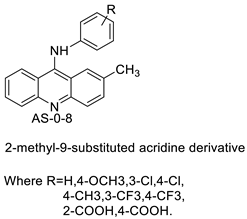 |
Lung carcinoma, breast cancer. |
[44] |
| CK0403 | 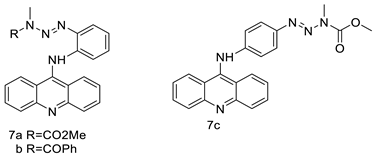 |
Anti-cancer activity | [45] |
| New series of 9-anilinoacridines containing phenyl-urea moieties | 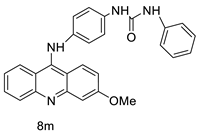 |
Novel dual Src and MEK inhibitors. | [46] |
| BO-1051 | 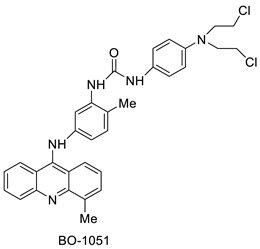 |
Against oral cancer | [47] |
| Novel isoxazole substituted 9 –anilinoacridines | 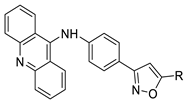 |
HER2 inhibitors. | [48] |
2.4. Acridine Thiourea Gold
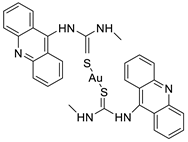
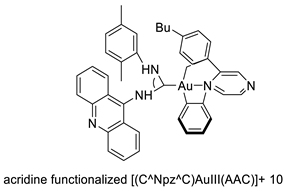
2.5. Acridine-Thiazolidinone
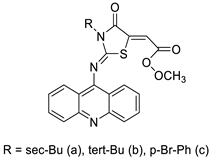
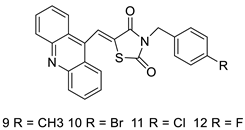
2.6. Acridinone
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Aseries of acridinones inspired by the structure of podophyllo toxin | 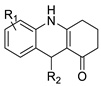 |
Triple-negative breast cancer cell line MDA-MB-231 | [53] |
| A new series of 9(10H)-acridinone-1,2,3 triazole derivatives | 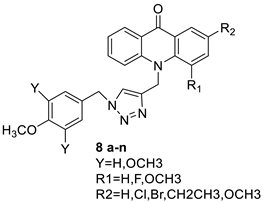 |
Against human breast cancer cell lines. | [54] |
| C-1305 C-1311 |
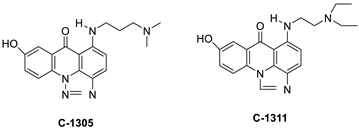 |
Anti-tumour agents | [55] |
2.7. Benzimidazole Substituted Acridines
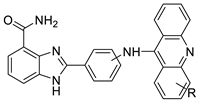
2.8. Benzoacridine
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Tubulin polymerisation inhibitors |  |
Eight cancer cell lines including MCF-7, A2780, HeLa, HepG2, DU145, A549, PC3, and LNCAP | [59] |
| New 7-substituted-5, 6-dihydrobenzo[c]acridine derivatives | 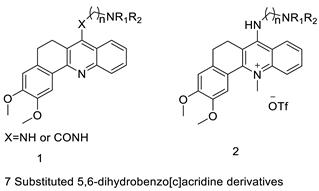 |
Induce apoptosis through activation of the caspase-3 cascade pathway | [60] |
| Phenanthrene fused-tetra hydrodibenzo-acridinones. | 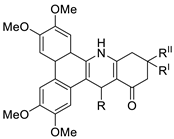 |
Cervical (HeLa), prostate (PC-3), fibrosarcoma (HT-1080), ovarian (SKOV-3) cancer cells | [61] |
| 12-arylbenzoacridines | 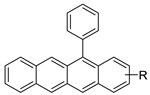 |
Selective estrogen-receptor modulators (SERMs). | [62] |
2.9. Imidazoacridinone
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
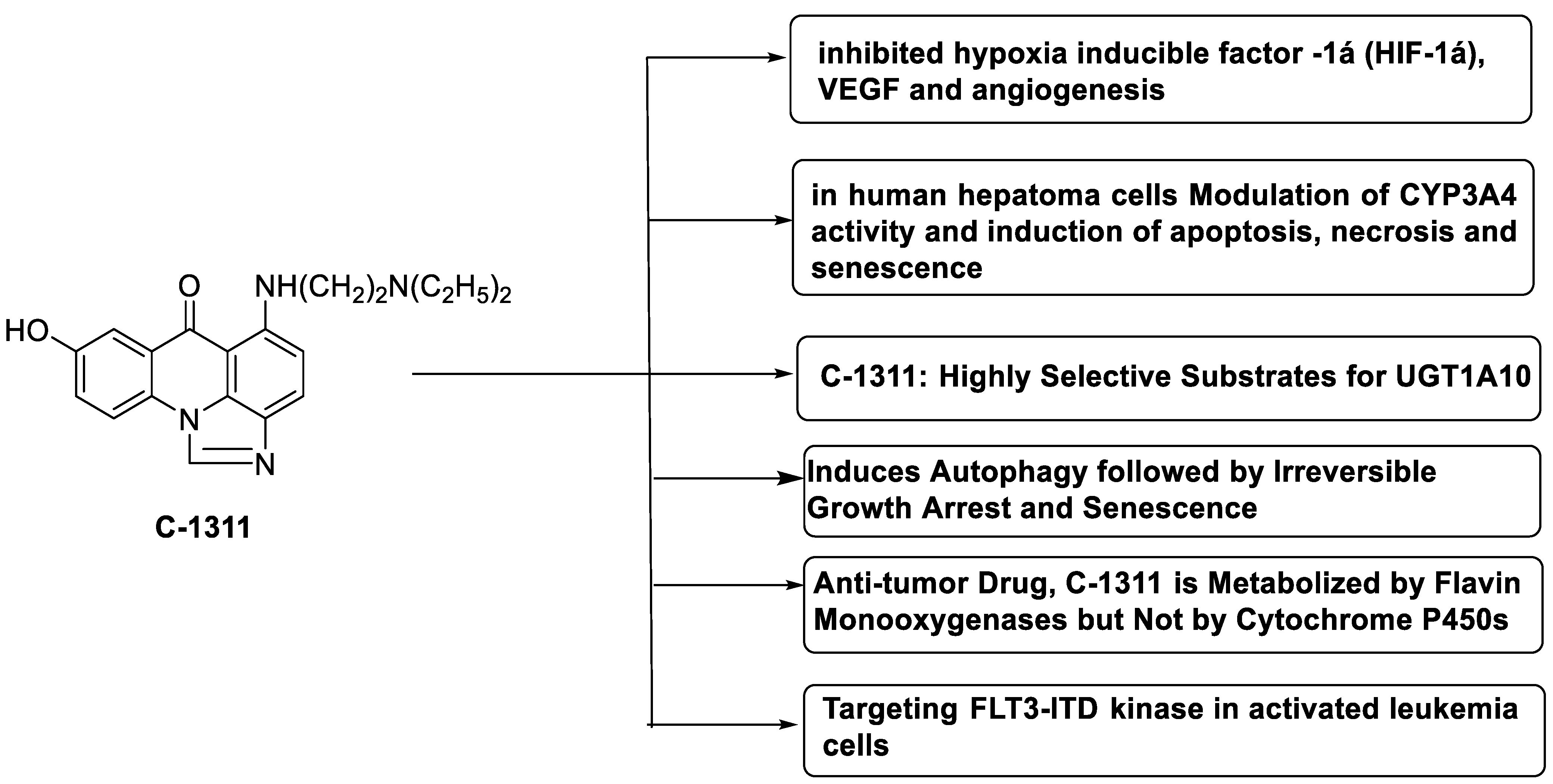
| Imidazoacridinone C-1311 | 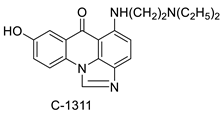 |
Anti-cancer activity | [63] |
| A novel photo activation-based pharmacological Trojan horse approach | 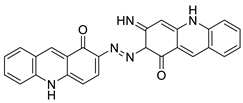 |
MDR tumour cells | [64] |
| C-1311 | 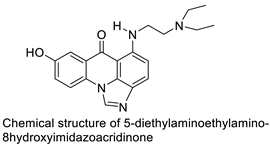 |
Anti-tumour agent C-1311in hepatoma cells | [65] |
| Crystal structures of NQO2 containing two of the imidazoacridin-6-ones | 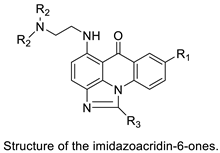 |
Inhibits the enzymatic function of NQO2 in cells. | [66] |
| Imidazoacridinone and triazoloacridinone | 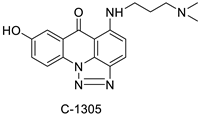 |
Liver cancer | [67] |
| C-1311 | 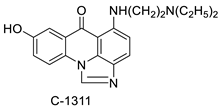 |
Human non-small-cell lung cancer (NSCLC) cell lines, A549 and H460. In A549 cells | [68] |
| C-1311 and C-1330 | 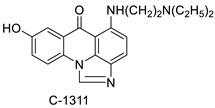 |
Human live microsomal enzymes | [69] |
| Imidazoacridinone C-1311 | 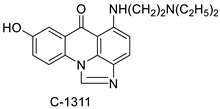 |
Against FLT3-ITD kinase | [70] |
2.10. Nitroacridine
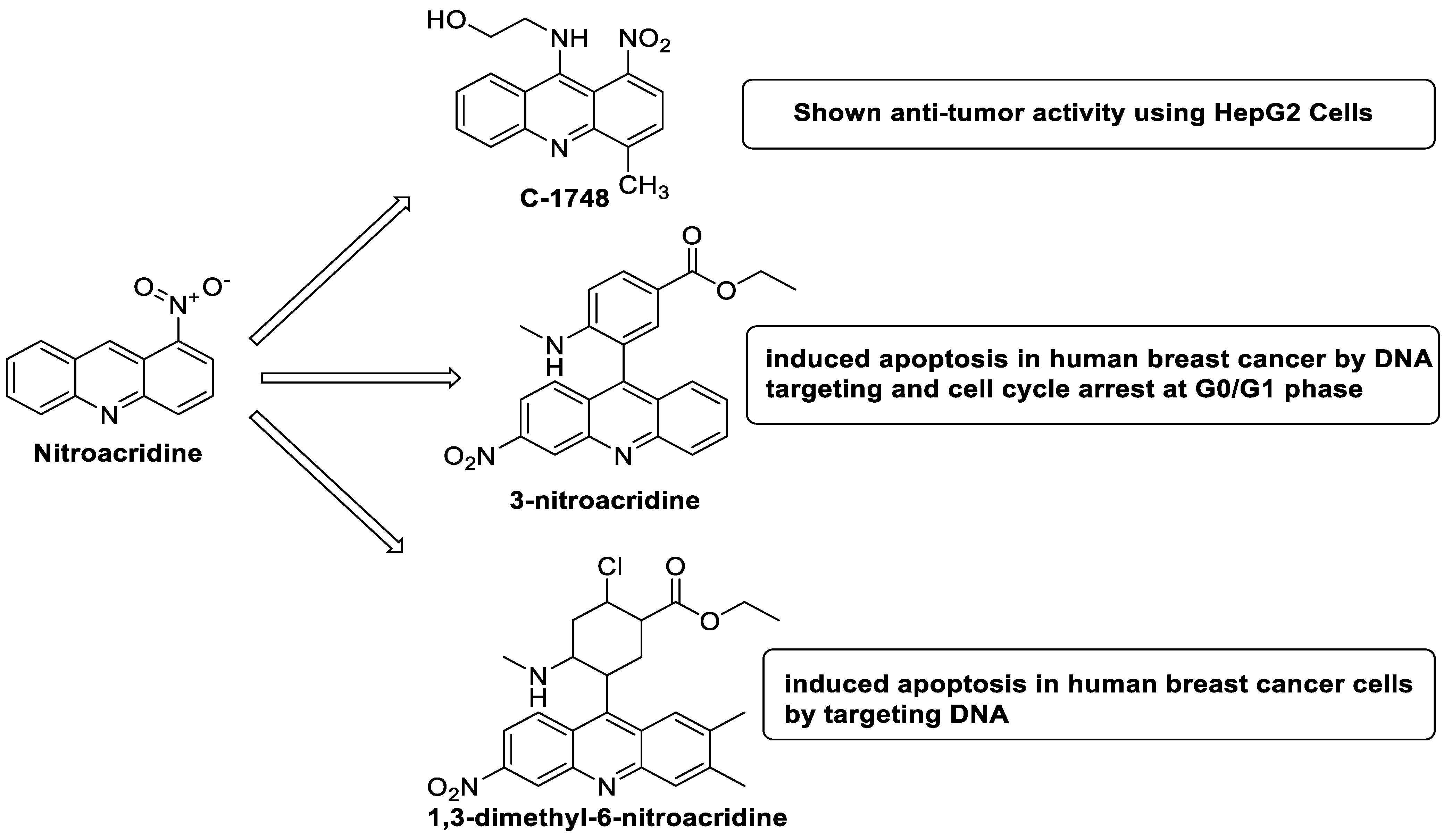
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| C-1748, 9-(20-hydroxyethylamino)-4-methyl-1-nitroacridine | 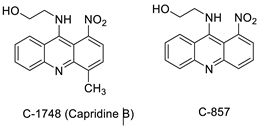 |
Anti-tumour activity | [71] |
| 3-nitroacridine derivatives | 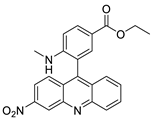 |
DNA-binding affinity and good inhibitory effect of tumour cell proliferation | [72] |
| New series of 1,3-dimethyl-6-nitroacridine derivatives | 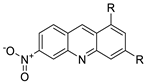 |
Anti-cancer activity against four cancer cells | [73] |
2.11. Oxazine
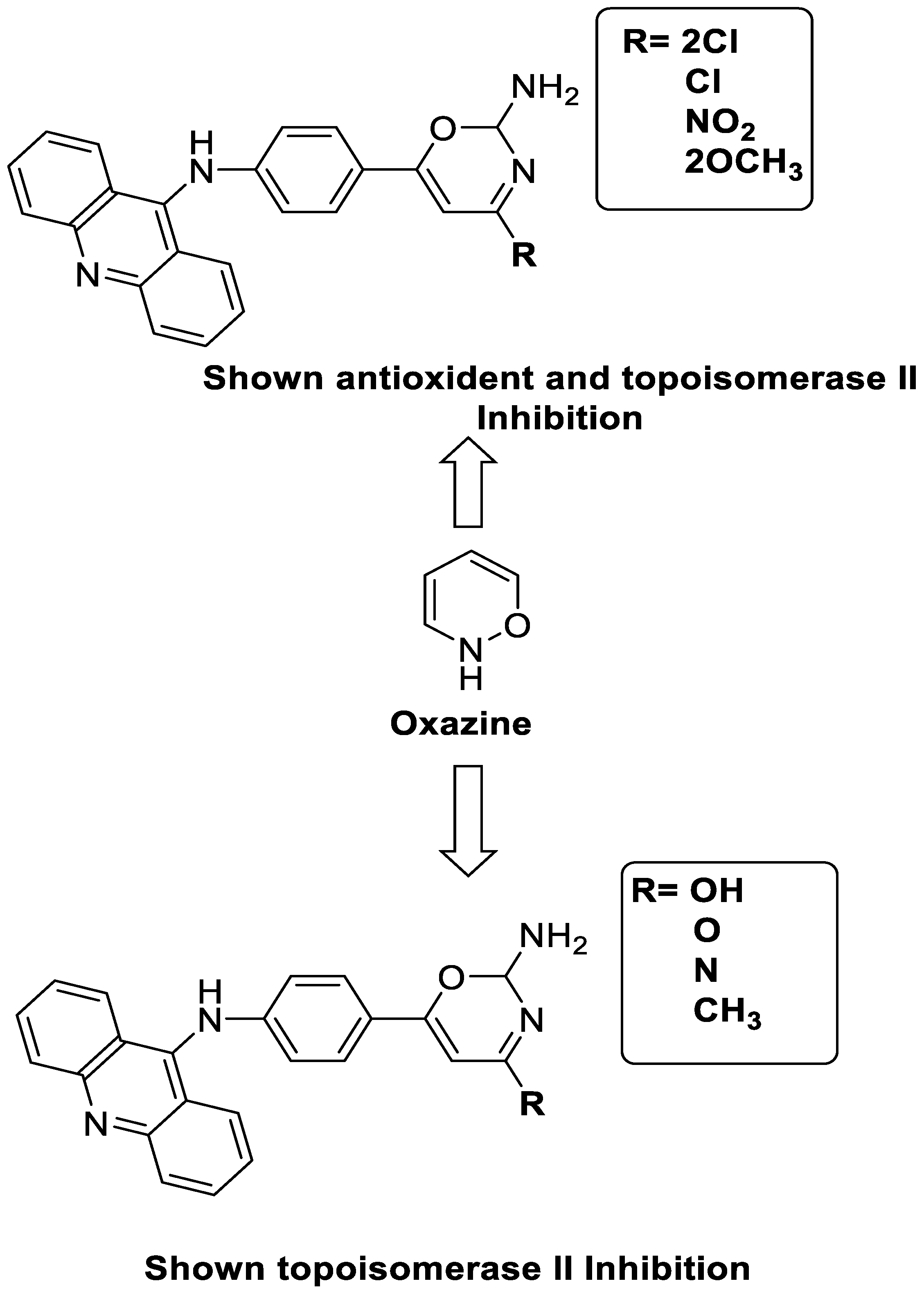
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| A series of 9-anilinoacridines substituted with oxazine derivatives | 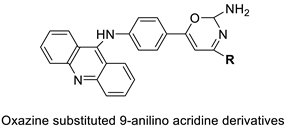 |
Antioxidant and anti-cancer activity | [74] |
| Novel oxazine substituted 9-anilinoacridines | 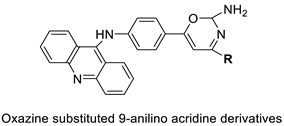 |
Topoisomerase II | [75] |
| A series of oxazine substituted 9-anilinoacridines | 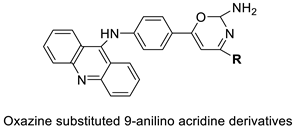 |
Anti-tumour activity on Dalton’s lymphoma ascites cells | [76] |
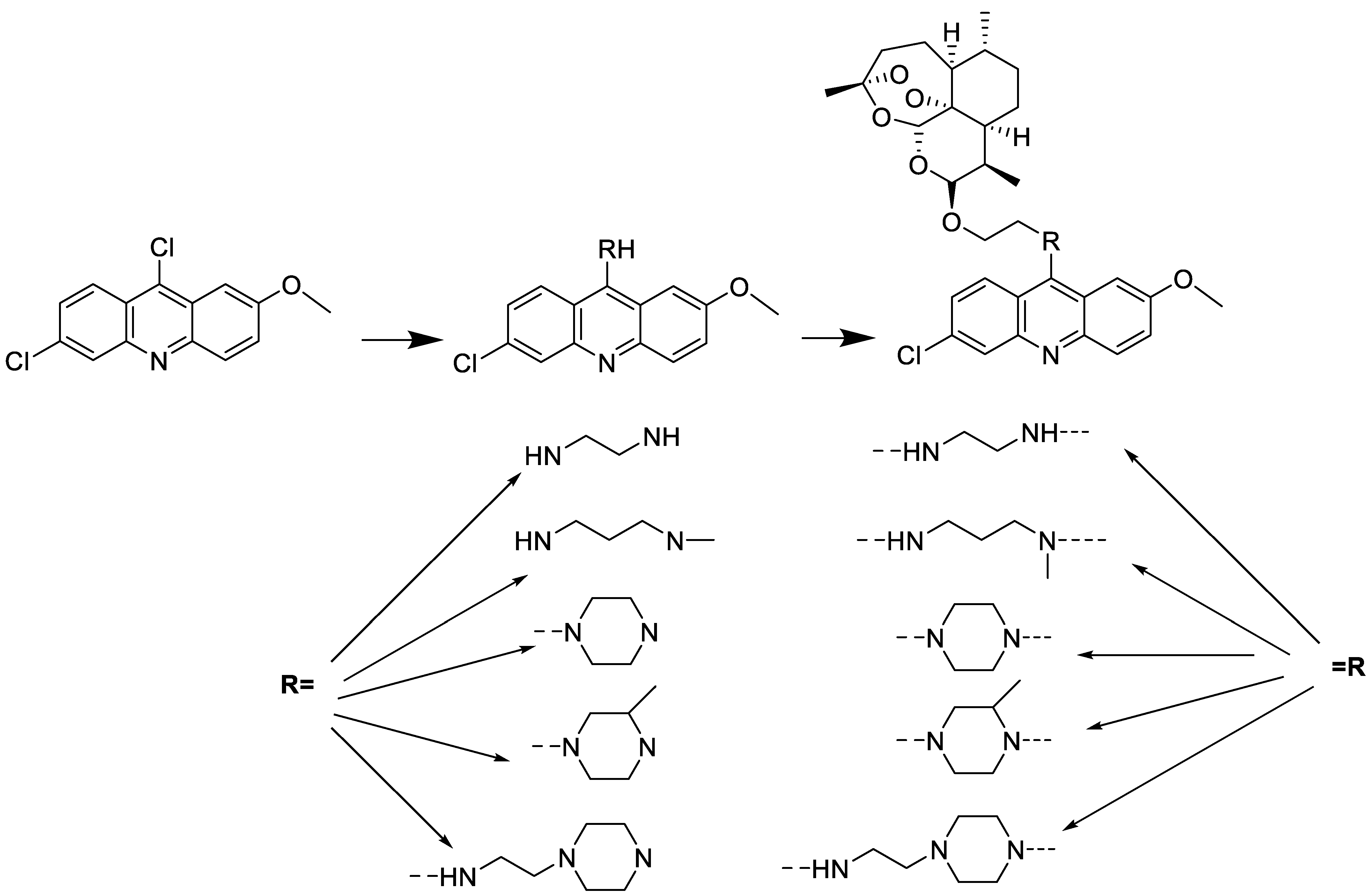
2.12. Platinum-Acridine Anti-Cancer Agents
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Non-classical platinum-acridine hybrid agents | 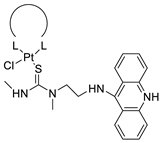 |
Distinct modules of DNA repair | [77] |
| A novel pharmacophore comprising a DNA-targeted platinum–acridine hybrid agent | 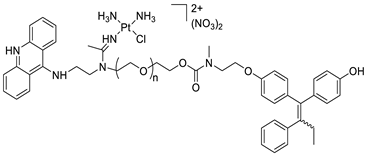 |
Breast cancer cell lines | [78] |
| P3A1 | 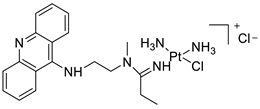 |
Cancer cells using MWCNTs as a drug carrier | [79] |
| [Pt(en)(L/L0)](NO3)2 [PtCl(en)(LH/L0H)](NO3)2 |
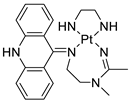 |
Inhibits H460 lung cancer cell proliferation | [80] |
| Platinum−acridine hybrid agents | 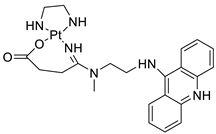 |
Anti-cancer activity | [81] |
| Platinum acridine hybrid agent [PtCl(en)(L)](NO3)2 | 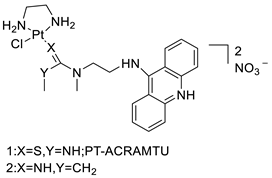 |
Delineate mechanistic differences between the platinum acridine hybrid agent | [82] |
| 7-aminobenz[c] acridine | 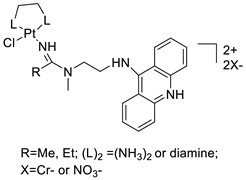 |
Tested in five NSCLC cell lines | [83] |
| Platinum–acridine agent | 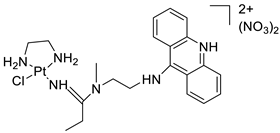 |
Structurally characterised | [84] |
| Platinum-acridine anti-cancer agent [PtCl(en)(LH)](NO3)2 | 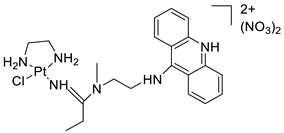 |
Chemo resistant non-small-cell lung cancer (NSCLC) cell lines | [85] |
2.13. Quinacrine
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Quinacrine | 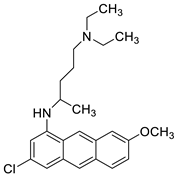 |
Human leukaemia K562cells | [86] |
| Quinacrine | 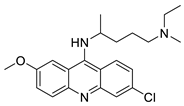 |
Human leukaemia U937 cells. | [87] |
| curcumin (Cur) and quinacrine (QC) | 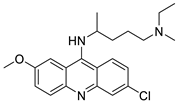 |
Induced breast epithelial transformed (MCF-10A-Tr) generated metastatic cells | [88] |
| Quinacrine | 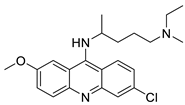 |
Inhibition of PARP-1PARylation | [89] |
| Quinacrine | 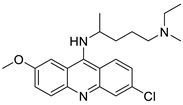 |
Cytotoxic effect of QC on DLBCL cells | [90] |
| Quinacrine | 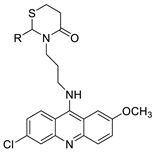 |
Against different cancer cell types | [91][92] |
2.14. Thiazacridine
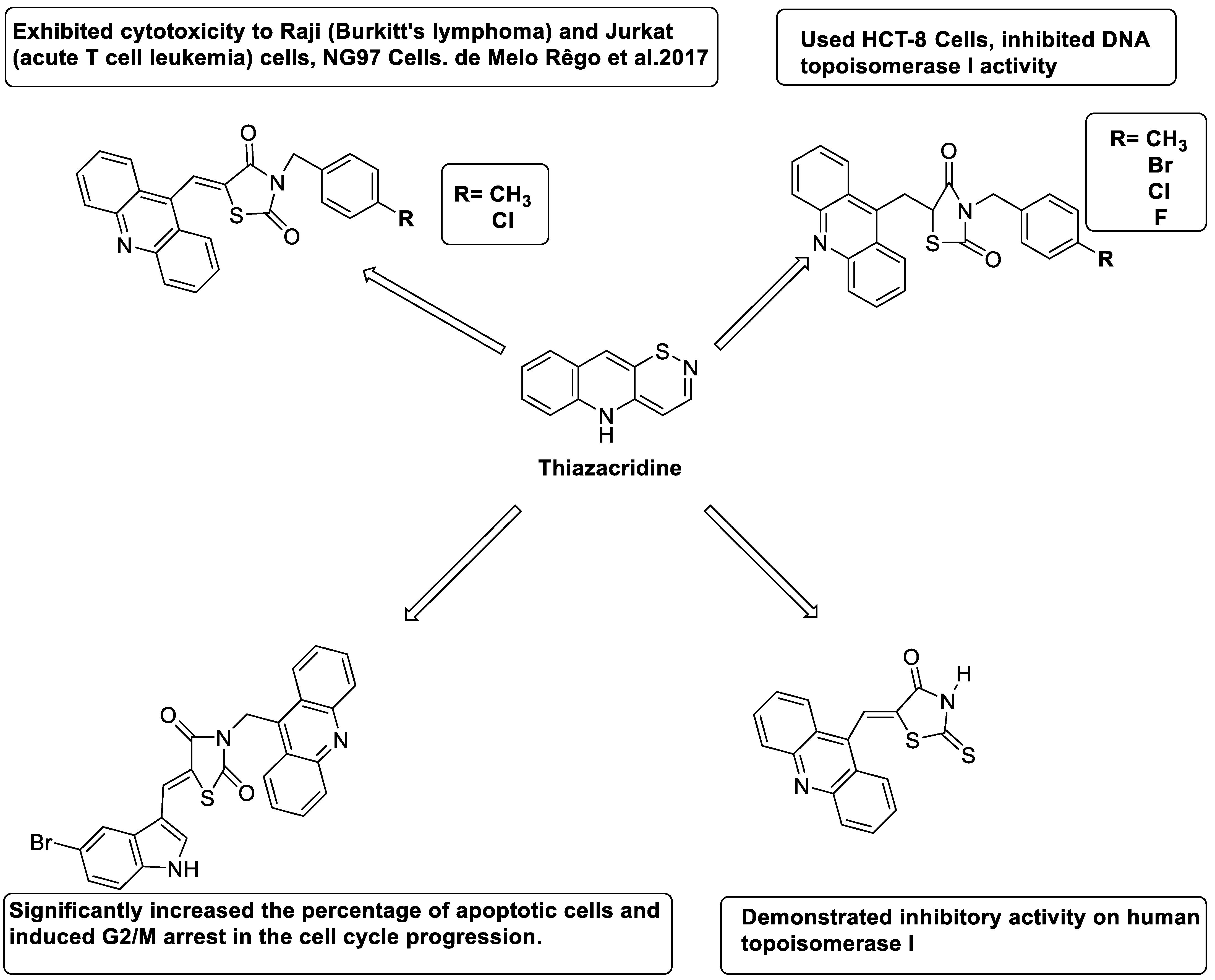
| Compound | Structure | Biological Activity | Reference |
|---|---|---|---|
| Three new thiazacridine derivatives | 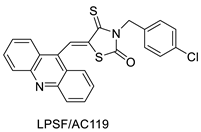 |
Induced neoplastic cell death primarily by apoptosis | [93] |
| An acridine and thiazolidine nucleus | 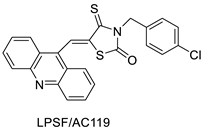 |
Tested in human colon carcinoma HCT-8 cells | [94] |
| Series of new thiazacridine agents | 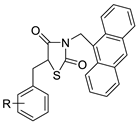 |
HepG2 cells | [95] |
| Thiazacridine and imidazacridine derivatives | 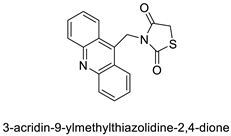 |
Tumours suppressors in some cancer cell lines | [96] |
2.15. Azacridine

| Potent Compound | Cancer Cell Lines | Control | IC50 (µM) | Reference |
|---|---|---|---|---|
| Propyl-AcrDTU | HL-60 | - | 7.2–8 | [1] |
| MAnT | HeLa cervical cancer cell line | Tetrandrine | 2.74 | [2] |
| C-1748 | HT29 Cell | Sorafenib | 39.7 | [3] |
| Cmpd-1 | A549 Cells | Etoposide | 15 | [4] |
| Cmpd-2 | A549 Cells | “ | 10 | |
| Cmpd-3 | A549 Cells | “ | 15 | |
| Cmpd-4 | A549 Cells | “ | 10 | |
| ACSO3 | HCT-116 Cells | Doxorubicin | 23.11 ± 1.03 | [7] |
| HeLa | “ | ˃50 | ||
| MCF-7 | “ | ˃50 | ||
| K562 | “ | ˃50 | ||
| HL-60 | “ | ˃50 | ||
| HaCat | “ | 62.18 ± 1.15 | ||
| PBMC | “ | 115.2 ± 5.82 | ||
| Cisplatin | MCF-7 | - | 12.7 ± 2.2 | [9] |
| 9-Acridine carboxylic acid | “ | - | 21.5 ± 3.1 | |
| Eu(III)-complex | “ | - | 14.3 ± 2.6 | |
| C-1748 | MiaPaCa-2 | - | 0.015 | [10] |
| AsPC-1 | - | 0.075 | ||
| B1 | SAS Cells | - | 1.5 | [13] |
| B2 | “ | - | 11.7 | |
| B3 | “ | - | 5.3 | |
| B4 | “ | - | 18.0 | |
| B5 | “ | - | 9.4 | |
| 80c | U937 HCT-116 |
m-AMSA | 0.90 | [14] |
| LS-1-10 | DLD1 Cells | Amsacrine | 1.31 | [15] |
| ACPH | A375 | Camptothecin | 20.74 | [18] |
| 1i | A549 HT-29 |
Etoposide | 59.12–14.87 17.32–5.90 |
[19] |
| 4b,4d,4e | HeLa, HepG2, HEK 239 |
Camptothecin | 18.89–52.64 18.7–108.34 31.38–277.13 |
[20] |
| C2 | malignant glioma and other human cancers. | erlotinib | 0.75–5 | [24] |
| C-1311 | DU-145 HCT-116 MBA-MB-231 |
Gemcitabine Erlotinib |
0.01–0.03 0.01–0.03 0.01–0.03 |
[25] |
| 4b 4f 4g 4i 4j |
HepG2 MCF-7 |
Doxorubicin | 1.4 ± 0.11 4.7 ± 0.09 2.2 ± 0.09 5.3 ± 0.16 4.8 ± 0.12 4.4 ± 0.10 2.6 ± 0.11 5.9 ± 0.15 1.6 ± 0.14 5.0 ± 0.18 |
[26] |
| SR374 SR375 SR361 SR362 |
MCF7 A549 GIST48 WI38 |
- | 1.6, 0.4, 2.9, <0.1 0.3, 0.1, 0.5, 0.1 1.5, 1.6, 2.8, 2.3 7.4, 4.5, 10.3, 0.7 |
[28] |
| Cmpd 5 | HL-60 | Acridine | 25.87 32.16 |
[29] |
| Cmpd 13 Cmpd 15 Cmpd 16 Cmpd 17 Cmpd 18 |
HT-29 | BRACO19 | 13.17–12.55 41.12–27.38 69.99–22.12 48.80–23.25 44.89–33.23 |
[31] |
| CuGGHK-Acr GGHK-Acr Ho-Acr |
HuH-7 MCF-7 Caco2 |
- | 9.8 ± 2.3 26.6 ± 2.2 16.7 ± 1.6 |
[33] |
| 8p | A549 Cells | Doxorubicin Etoposide Amsacrine |
0.61 ± 0.06 | [34] |
| 9AmAcPtCl2 | HeLa Cells | Cisplatin | 0.4 | [40] |
| 7r | HepG-2 MCF-7 |
Colchicin | 4.2 23.8 |
[41] |
| 7b | H1299 WM264 HCT116 |
Cisplatin | 2.9 0.8 15.4 |
[45] |
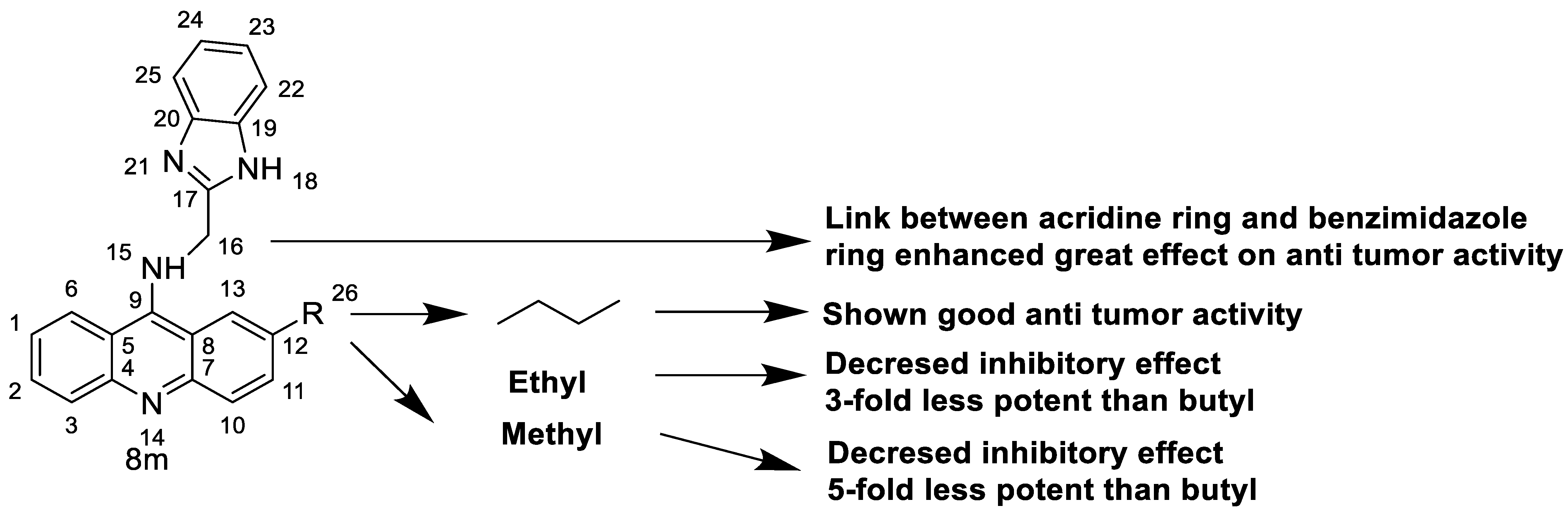
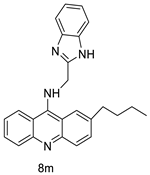
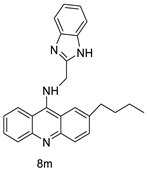
| 8m | K562 HepG-2 |
Imatinib | 4.08 ± 0.14 9.41 ± 1.09 |
[46] |
| BO-1051 | SAS Cells OECM1 |
- | 2.39 1.97 |
[47] |
| Cmpd 2 | A2780 MDA-MB-231 SK-BR-3 MCF-7 |
CDDP AuC1PPh3 |
0.88 ± 0.20 2.75 ± 0.40 3.62 ± 0.14 5.32 ± 0.65 |
[50] |
| Complex 11 | A549 MCF-7 HL60 |
Cisplatin | 7.6 ± 0.6 1.5 ± 0.1 1.1 ± 0.1 |
[50] |
| 2C | HL-60 L1210 A2780 |
Cisplatin | 1.3 ± 0.2 3.1 ± 0.4 7.7 ± 0.5 |
[51] |
| Cmpd-9 | PC-3 COLO-205 |
Amsacrine | 5.5–9.5 8.6–42.3 |
[52] |
| Cmpd-6 Cmpd-7 Cmpd-9 Cmpd-10 |
MDA-MB-231 DU-145 |
Colchicine | 3 ± 1, 3.2 ± 0.7 0.190 ± 0.007 1.1 ± 0.2 1.0 ± 0.3 0.11 ± 0.02 0.12 ± 0.02 |
[53] |
| 8c | MCF-7 T-47D MDA-MB-231 |
etoposide | 11 ± 4.8 14.5 ± 5.2 16.6 ± 5.9 |
[54] |
| C-1305 C-1311 |
KB-3 | - | 0.263 ± 0.016 0.106 ± 0.014 |
[55] |
| 8m | SW480 HCT116 |
- | 6.77 ± 0.19 3.33 ± 0.02 |
[56] |
| 11L | PARP-1 MCF-7 |
Olaparib m-AMSA |
0.45 ± 0.03 2.14 ± 0.92 |
[57] |
| 8I | K562 HepG-2 |
Colchicine Imatinib |
2.68 8.11 |
[58] |
| 4g | HUVEC LNCAP |
Β-Lapachone | 49.19 ± 2.3 18.54 ± 2.11 |
[59] |
| 2b | HeLa K562 A549 |
- | 3.2 9.2 5.7 |
[60] |
| 8m | SKOV-3 | Cisplatin | 0.24 ± 0.05 | [61] |
| Cmpd-1 | MCF-7 | Tamoxifen citrate | 0.951 | [62] |
| 6a1 | HCT116 | NQO2 | 14 ± 4 | [66] |
| C-1311 | MV4-11 MOLM13 |
- | 0.03 ± 0.01 0.04 ± 0.02 |
[70] |
| Cmpd-2 | MCF-7 MDA-MB-231 SGC7901 MGC803 |
- | 6.79 ± 2.01 6.46 ± 2.19 17.25 ± 3.84 10.94 ± 2.26 |
[72] |
| Cmpd-1 Cmpd-6 |
MCF-7 MDA-MB-231 SGC7901 MGC803 |
- | 8.83 ± 0.98 10.02 ± 0.78 41.47 ± 3.24 23.96 ± 1.54 |
[73] |
| 5a | DLA Cells | ascorbic acid | 20.03 ± 0.2583 | [74] |
| 8 | MCF-7 MDA-MB-231 |
Tamoxifen | 1.6 ± 0.4 13.2 ± 0.1 |
[78] |
| Cmpd-3 | NCI-H460 OVCAR-3 PANC-1 |
Cisplatin | 0.008 ± 0.002 1.1 ± 0.1 0.09 ± 0.01 |
[80] |
| 3a 3b |
H460 | - | 12 ± 2 2.8 ± 0.3 |
[81] |
| P1-A1 P1-B1 P1-B2 |
NCI-H460 | - | 0.0052 ± 0.0001 0.24 ± 0.01 2.4 ± 0.5 |
[83] |
| Cmpd-1 | NCI-H460 NCI-H522 |
Cisplatin | 8 ± 2 18 ± 2 |
[85] |
| QC | OCI-Ly01 SU-DHL-8 |
Quinacrine | 1.8 2 |
[90] |
| Cmpd-25 | MDA-MB-468 MDA-MB-231 MCF-7 184B5 |
Cisplatin Quinacrine |
1.73 ± 0.80 2.80 ± 1.30 0.69 ± 0.41 4.96 ± 0.24 |
[91] |
| Cmpd-11 | MDA-MB-468 MDA-MB-231 MCF-7 184B5 |
Cisplatin Quinacrine |
2.40 ± 1.01 1.92 ± 0.20 1.24 ± 0.51 16.16 ± 0.81 |
[92] |
| LPSF/AC-119 | Raji Jurkat |
Amsacrine | 0.6 1.53 |
[93] |
| AC-4 AC-7 AC-10 AC-23 |
HCT-8 | Amsacrine | 3.1 5.3 3.6 2.3 |
[94] |
| 7a 7b |
HepG2 | Amsacrine | 68.03 48.63 |
[95] |
| 13b | K562 A549 |
Imatinib | 0.22 ± 0.03 0.253 ± 0.16 |
[97] |
This entry is adapted from the peer-reviewed paper 10.3390/molecules28010193
References
- Cisáriková, A.; Barbieriková, Z.; Janovec, L.; Imrich, J.; Hunáková, L.; Bačová, Z.; Paulíková, H. Acridin-3, 6-dialkyldithiourea hydrochlorides as new photosensitizers for photodynamic therapy of mouse leukaemia cells. Bioorg. Med. Chem. 2016, 24, 2011–2022.
- Calvillo-Páez, V.; Sotelo-Mundo, R.R.; Leyva-Peralta, M.; Gálvez-Ruiz, J.C.; Corona-Martínez, D.; Moreno-Corral, R.; Escobar-Picos, R.; Höpfl, H.; Juárez-Sánchez, O.; Lara, K.O. Synthesis, spectroscopic, physicochemical and structural characterization of tetrandrine-based macrocycles functionalized with acridine and anthracene groups: DNA binding and anti-proliferative activity. Chem.-Biol. Interact. 2018, 286, 34–44.
- Mróz, A.; Ryska, I.; Sominko, H.; Bejrowska, A.; Mazerska, Z. Drug-drug interaction potential of antitumor acridine agent C-1748: The substrate of UDP-glucuronosyltransferases 2B7, 2B17 and the inhibitor of 1A9 and 2B7. Pharmacol. Rep. 2018, 70, 972–980.
- Szymański, P.; Olszewska, P.; Mikiciuk-Olasik, E.; Różalski, A.; Maszewska, A.; Markiewicz, L.; Cuchra, M.; Majsterek, I. Novel tetrahydroacridine and cyclopentaquinoline derivatives with fluorobenzoic acid moiety induce cell cycle arrest and apoptosis in lung cancer cells by activation of DNA damage signaling. Tumor Biol. 2017, 39, 1010428317695011.
- Takac, P.; Kello, M.; Vilkova, M.; Vaskova, J.; Michalkova, R.; Mojzisova, G.; Mojzis, J. Antiproliferative effect of acridine chalcone is mediated by induction of oxidative stress. Biomolecules 2020, 10, 345.
- Silva, D.K.F.; Duarte, S.S.; Lisboa, T.M.; Ferreira, R.C.; Lopes, A.L.d.; Carvalho, D.; Rodrigues-Mascarenhas, S.; da Silva, P.M.; Segundo, M.A.; de Moura, R.O. Antitumor effect of a novel spiro-acridine compound is associated with up-regulation of Th1-type responses and antiangiogenic action. Molecules 2020, 25, 29.
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M. Toxicity and antitumor activity of a thiophene–acridine hybrid. Molecules 2020, 25, 64.
- Almeida, S.M.V.; Silva, L.P.B.G.; Lima, L.R.A.; Botelho, S.P.S.; Lima, M.d.C.; Pitta, I.d.; Beltrão, E.I.C.; Junior, L.B.C. Dimethyl-2--malonate as fluorescent probe for histochemical analysis. Microsc. Res. Tech. 2017, 80, 608–614.
- Azab, H.A.; Hussein, B.H.; El-Azab, M.F.; Gomaa, M.; El-Falouji, A.I. Bis (acridine-9-carboxylate)-nitro-europium (III) dihydrate complex a new apoptotic agent through Flk-1 down regulation, caspase-3 activation and oligonucleosomes DNA fragmentation. Bioorg. Med. Chem. 2013, 21, 223–234.
- Borowa-Mazgaj, B.; Mróz, A.; Augustin, E.; Paluszkiewicz, E.; Mazerska, Z. The overexpression of CPR and P450 3A4 in pancreatic cancer cells changes the metabolic profile and increases the cytotoxicity and pro-apoptotic activity of acridine antitumor agent, C-1748. Biochem. Pharmacol. 2017, 142, 21–38.
- Campbell, A.P.; Wakelin, L.P.; Denny, W.A.; Finch, A.M. Homobivalent conjugation increases the allosteric effect of 9-aminoacridine at the α1-adrenergic receptors. Mol. Pharmacol. 2017, 91, 135–144.
- Carvalho, J.; Lopes-Nunes, J.; Lopes, A.C.; Campello, M.P.C.; Paulo, A.; Queiroz, J.A.; Cruz, C. Aptamer-guided acridine derivatives for cervical cancer. Org. Biomol. Chem. 2019, 17, 2992–3002.
- Chai, H.; Hazawa, M.; Hosokawa, Y.; Igarashi, J.; Suga, H.; Kashiwakura, I. Novel acridine-based N-acyl-homoserine lactone analogs induce endoreduplication in the human oral squamous carcinoma cell line SAS. Biol. Pharm. Bull. 2012, 35, 1257–1263.
- Chen, J.; Li, D.; Li, W.; Yin, J.; Zhang, Y.; Yuan, Z.; Gao, C.; Liu, F.; Jiang, Y. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg. Med. Chem. 2018, 26, 3958–3966.
- Fu, W.; Li, X.; Lu, X.; Zhang, L.; Li, R.; Zhang, N.; Liu, S.; Yang, X.; Wang, Y.; Zhao, Y. A novel acridine derivative, LS-1-10 inhibits autophagic degradation and triggers apoptosis in colon cancer cells. Cell Death Dis. 2017, 8, e3086.
- Gardette, M.; Papon, J.; Bonnet, M.; Desbois, N.; Labarre, P.; Wu, T.-D.; Miot-Noirault, E.; Madelmont, J.-C.; Guerquin-Kern, J.-L.; Chezal, J.-M. Evaluation of new iodinated acridine derivatives for targeted radionuclide therapy of melanoma using 125I, an Auger electron emitter. Investig. New Drugs 2011, 29, 1253–1263.
- Gardette, M.; Viallard, C.; Paillas, S.; Guerquin-Kern, J.-L.; Papon, J.; Moins, N.; Labarre, P.; Desbois, N.; Wong-Wah-Chung, P.; Palle, S. Evaluation of two 125I-radiolabeled acridine derivatives for Auger-electron radionuclide therapy of melanoma. Investig. New Drugs 2014, 32, 587–597.
- Ghosh, R.; Bhowmik, S.; Guha, D. 9-phenyl acridine exhibits antitumour activity by inducing apoptosis in A375 cells. Mol. Cell. Biochem. 2012, 361, 55–66.
- Girek, M.; losiński, K.K.; Grobelski, B.; Pizzimenti, S.; Cucci, M.A.; Daga, M.; Barrera, G.; Pasieka, Z.; Czarnecka, K.; Szymański, P. Novel tetrahydroacridine derivatives with iodobenzoic moieties induce G0/G1 cell cycle arrest and apoptosis in A549 non-small lung cancer and HT-29 colorectal cancer cells. Mol. Cell. Biochem. 2019, 460, 123–150.
- Haider, M.R.; Ahmad, K.; Siddiqui, N.; Ali, Z.; Akhtar, M.J.; Fuloria, N.; Fuloria, S.; Ravichandran, M.; Yar, M.S. Novel 9-(2-(1-arylethylidene) hydrazinyl) acridine derivatives: Target Topoisomerase 1 and growth inhibition of HeLa cancer cells. Bioorg. Chem. 2019, 88, 102962.
- Hansda, S.; Ghosh, G.; Ghosh, R. 9-phenyl acridine photosensitizes A375 cells to UVA radiation. Heliyon 2020, 6, e04733.
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J. Acridine derivatives as inhibitors of the ire1α–xbp1 pathway are cytotoxic to human multiple myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065.
- Mangueira, V.M.; Batista, T.M.; Brito, M.T.; de Sousa, T.K.G.; da Cruz, R.M.D.; de Abrantes, R.A.; Veras, R.C.; de Medeiros, I.A.; Medeiros, K.K.d.; Pereira, A.L.d. A new acridine derivative induces cell cycle arrest and antiangiogenic effect on Ehrlich ascites carcinoma model. Biomed. Pharmacother. 2017, 90, 253–261.
- Qi, Q.; He, K.; Yoo, M.-H.; Chan, C.-B.; Liu, X.; Zhang, Z.; Olson, J.J.; Xiao, G.; Wang, L.; Mao, H. Acridine yellow G blocks glioblastoma growth via dual inhibition of epidermal growth factor receptor and protein kinase C kinases. J. Biol. Chem. 2012, 287, 6113–6127.
- Paluszkiewicz, E.; Horowska, B.; Borowa-Mazgaj, B.; Peszyńska-Sularz, G.; Paradziej-Lukowicz, J.; Augustin, E.; Konopa, J.; Mazerska, Z. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020, 204, 112599.
- Ramesh, K.B.; Pasha, M.A. Study on one-pot four-component synthesis of 9-aryl-hexahydro-acridine-1, 8-diones using SiO2–I as a new heterogeneous catalyst and their anticancer activity. Bioorg. Med. Chem. Lett. 2014, 24, 3907–3913.
- Raza, A.; Jacobson, B.A.; Benoit, A.; Patel, M.R.; Jay-Dixon, J.; Hiasa, H.; Ferguson, D.M.; Kratzke, R.A. Novel acridine-based agents with topoisomerase II inhibitor activity suppress mesothelioma cell proliferation and induce apoptosis. Investig. New Drugs 2012, 30, 1443–1448.
- Roe, S.; Gunaratnam, M.; Spiteri, C.; Sharma, P.; Alharthy, R.D.; Neidle, S.; Moses, J.E. Synthesis and biological evaluation of hybrid acridine-HSP90 ligand conjugates as telomerase inhibitors. Org. Biomol. Chem. 2015, 13, 8500–8504.
- Salem, O.M.; Vilková, M.; JanoĿková, J.; Jendželovskỳ, R.; FedoroĿko, P.; Žilecká, E.; Kašpárková, J.; Brabec, V.; Imrich, J.; Kožurková, M. New spiro tria (thia) zolidine acridines as topoisomerase inhibitors, DNA binders and cytostatic compounds. Int. J. Biol. Macromol. 2016, 86, 690–700.
- Singh, P.; Kumar, A.; Sharma, A.; Kaur, G. Identification of amino acid appended acridines as potential leads to anti-cancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3854–3858.
- Ungvarsky, J.; Plsikova, J.; Janovec, L.; Koval, J.; Mikes, J.; Mikesová, L.; Harvanova, D.; Fedorocko, P.; Kristian, P.; Kasparkova, J. Novel trisubstituted acridines as human telomeric quadruplex binding ligands. Bioorg. Chem. 2014, 57, 13–29.
- Wilks, T.R.; Pitto-Barry, A.; Kirby, N.; Stulz, E.; O’Reilly, R.K. Construction of DNA–polymer hybrids using intercalation interactions. Chem. Commun. 2014, 50, 1338–1340.
- Yu, Z.; Han, M.; Cowan, J.A. Toward the Design of a Catalytic Metallodrug: Selective Cleavage of G-Quadruplex Telomeric DNA by an Anticancer Copper–Acridine–ATCUN Complex. Angew. Chem. 2015, 127, 1921–1925.
- Zhang, W.; Zhang, B.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70.
- Ferguson, D.M.; Jacobson, B.A.; Jay-Dixon, J.; Patel, M.R.; Kratzke, R.A.; Raza, A. Targeting topoisomerase II activity in NSCLC with 9-aminoacridine derivatives. Anticancer Res. 2015, 35, 5211–5217.
- Sun, Y.-W.; Chen, K.-Y.; Kwon, C.-H.; Chen, K.-M. CK0403, a 9-aminoacridine, is a potent anti-cancer agent in human breast cancer cells. Mol. Med. Rep. 2016, 13, 933–938.
- Aguilera, R.F.; Artali, R.; Benoit, A.; Gómez, R.G.; Casadellà, R.E.i.; Ferguson, D.M.; Sham, Y.Y.; Mazzini, S. Structure and stability of human telomeric G-quadruplex with preclinical 9-amino acridines. PLoS ONE 2013, 8, E57701.
- Joubert, J.P.; Smit, F.J.; Plessis, L.D.; Smith, P.J.; N’Da, D.D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin–acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27.
- Ju, W.; Zhang, M.; Petrus, M.; Maeda, M.; Pise-Masison, C.A.; Waldmann, T.A. Combination of 9-aminoacridine with Campath-1H provides effective therapy for a murine model of adult T-cell leukaemia. Retrovirology 2014, 11, 43.
- Kava, H.W.; Murray, V. CpG methylation increases the DNA binding of 9-aminoacridine carboxamide Pt analogues. Bioorg. Med. Chem. 2016, 24, 4701–4710.
- Luan, X.; Gao, C.; Zhang, N.; Chen, Y.; Sun, Q.; Tan, C.; Liu, H.; Jin, Y.; Jiang, Y. Exploration of acridine scaffold as a potentially interesting scaffold for discovering novel multi-target VEGFR-2 and Src kinase inhibitors. Bioorganic Med. Chem. 2011, 19, 3312–3319.
- Paul, M.; Murray, V. The sequence selectivity of DNA-targeted 9-aminoacridine cisplatin analogues in a telomere-containing DNA sequence. JBIC J. Biol. Inorg. Chem. 2011, 16, 735–743.
- Ryan, E.; Blake, A.J.; Benoit, A.; David, M.F.; Robert, A.K. Efficacy of substituted 9-aminoacridine derivatives in small cell lung cancer. Investig. New Drugs 2013, 31, 285–292.
- Kumar, R.; Sharma, A.; Sharma, S.; Silakari, O.; Singh, M.; Kaur, M. Synthesis, characterization and antitumor activity of 2-methyl-9-substituted acridines. Arab. J. Chem. 2017, 10, S956–S963.
- Walunj, D.; Egarmina, K.; Tuchinsky, H.; Shpilberg, O.; Hershkovitz-Rokah, O.; Grynszpan, F.; Gellerman, G. Expedient synthesis and anticancer evaluation of dual-action 9-anilinoacridine methyl triazene chimeras. Chem. Biol. Drug Des. 2021, 97, 237–252.
- Cui, Z.; Li, X.; Li, L.; Zhang, B.; Gao, C.; Chen, Y.; Tan, C.; Liu, H.; Xie, W.; Yang, T. Design, synthesis and evaluation of acridine derivatives as multi-target Src and MEK kinase inhibitors for anti-tumor treatment. Bioorg. Med. Chem. 2016, 24, 261–269.
- Lo, W.-L.; Chu, P.-Y.; Lee, T.-H.; Su, T.-L.; Chien, Y.; Chen, Y.-W.; Huang, P.-I.; Tseng, L.-M.; Tu, P.-H.; Kao, S.-Y. A Combined DNA-affinic molecule and N-mustard alkylating agent has an anti-cancer effect and induces autophagy in oral cancer cells. Int. J. Mol. Sci. 2012, 13, 3277–3290.
- Kalirajan, R.; Pandiselvi, A.; Gowramma, B.; Balachandran, P. In-silico design, ADMET screening, MM-GBSA binding free energy of some novel isoxazole substituted 9-anilinoacridines as HER2 inhibitors targeting breast cancer. Curr. Drug Res. Rev. Former. Curr. Drug Abus. Rev. 2018, 11, 118–128.
- Perez, S.A.; de Haro, C.; Vicente, C.; Donaire, A.; Zamora, A.; Zajac, J.; Kostrhunova, H.; Brabec, V.; Bautista, D.; Ruiz, J. New acridine thiourea gold (I) anticancer agents: Targeting the nucleus and inhibiting vasculogenic mimicry. ACS Chem. Biol. 2017, 12, 1524–1537.
- Williams, M.R.; Bertrand, B.; Fernandez-Cestau, J.; Waller, Z.A.; O’Connell, M.A.; Searcey, M.; Bochmann, M. Acridine-decorated cyclometallated gold (III) complexes: Synthesis and anti-tumour investigations. Dalton Trans. 2018, 47, 13523–13534.
- Paulíková, H.; Vantová, Z.; Hunáková, L.; Čižeková, L.; Čarná, M.; Kožurková, M.; Sabolová, D.; Kristian, P.; Hamul’aková, S.; Imrich, J. DNA binding acridine–thiazolidinone agents affecting intracellular glutathione. Bioorg. Med. Chem. 2012, 20, 7139–7148.
- Barros, F.W.; Silva, T.G.; Pitta, M.G.d.; Bezerra, D.P.; Costa-Lotufo, L.V.; de Moraes, M.O.; Pessoa, C.; de Moura, M.A.F.; de Abreu, F.C.; de Lima, M.d.C.A. Synthesis and cytotoxic activity of new acridine-thiazolidine derivatives. Bioorg. Med. Chem. 2012, 20, 3533–3539.
- Magalhaes, L.G.; Marques, F.B.; da Fonseca, M.B.; Rogerio, K.R.; Graebin, C.S.; Andricopulo, A.D. Discovery of a series of acridinones as mechanism-based tubulin assembly inhibitors with anticancer activity. PLoS ONE 2016, 11, e0160842.
- Mohammadi-Khanaposhtani, M.; Safavi, M.; Sabourian, R.; Mahdavi, M.; Pordeli, M.; Saeedi, M.; Ardestani, S.K.; Foroumadi, A.; Shafiee, A.; Akbarzadeh, T. Design, synthesis, in vitro cytotoxic activity evaluation, and apoptosis-induction study of new 9 (10H)-acridinone-1, 2, 3-triazoles. Mol. Divers. 2015, 19, 787–795.
- Pawlowska, M.; Chu, R.; Fedejko-Kap, B.; Augustin, E.; Mazerska, Z.; Radominska-Pandya, A.; Chambers, T.C. Metabolic transformation of antitumor acridinone C-1305 but not C-1311 via selective cellular expression of UGT1A10 increases cytotoxic response: Implications for clinical use. Drug Metab. Dispos. 2013, 41, 414–421.
- Chen, K.; Chu, B.; Liu, F.; Li, B.; Gao, C.; Li, L.; Sun, Q.; Shen, Z.; Jiang, Y. New benzimidazole acridine derivative induces human colon cancer cell apoptosis in vitro via the ROS-JNK signaling pathway. Acta Pharmacol. Sin. 2015, 36, 1074–1084.
- Yuan, Z.; Chen, S.; Chen, C.; Chen, J.; Chen, C.; Dai, Q.; Gao, C.; Jiang, Y. Design, synthesis and biological evaluation of 4-amidobenzimidazole acridine derivatives as dual PARP and Topo inhibitors for cancer therapy. Eur. J. Med. Chem. 2017, 138, 1135–1146.
- Gao, C.; Li, B.; Zhang, B.; Sun, Q.; Li, L.; Li, X.; Chen, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and biological evaluation of benzimidazole acridine derivatives as potential DNA-binding and apoptosis-inducing agents. Bioorg. Med. Chem. 2015, 23, 1800–1807.
- Behbahani, F.S.; Tabeshpour, J.; Mirzaei, S.; Golmakaniyoon, S.; Tayarani-Najaran, Z.; Ghasemi, A.; Ghodsi, R. Synthesis and biological evaluation of novel benzo acridine-diones as potential anticancer agents and tubulin polymerization inhibitors. Arch. Der Pharm. 2019, 352, 1800307.
- Guo, Q.-L.; Su, H.-F.; Wang, N.; Liao, S.-R.; Lu, Y.-T.; Ou, T.-M.; Tan, J.-H.; Li, D.; Huang, Z.-S. Synthesis and evaluation of 7-substituted-5, 6-dihydrobenzo acridine derivatives as new c-KIT promoter G-quadruplex binding ligands. Eur. J. Med. Chem. 2017, 130, 458–471.
- Kumar, N.P.; Sharma, P.; Reddy, T.S.; Shankaraiah, N.; Bhargava, S.K.; Kamal, A. Microwave-assisted one-pot synthesis of new phenanthrene fused-tetrahydrodibenzo-acridinones as potential cytotoxic and apoptosis inducing agents. Eur. J. Med. Chem. 2018, 151, 173–185.
- Torikai, K.; Koga, R.; Liu, X.; Umehara, K.; Kitano, T.; Watanabe, K.; Oishi, T.; Noguchi, H.; Shimohigashi, Y. Design and synthesis of benzoacridines as estrogenic and anti-estrogenic agents. Bioorg. Med. Chem. 2017, 25, 5216–5237.
- Paradziej-Lukowicz, J.; Skwarska, A.; Peszyńska-Sularz, G.; Brillowska-Dąbrowska, A.; Konopa, J. Anticancer imidazoacridinone C-1311 inhibits hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF) and angiogenesis. Cancer Biol. Ther. 2011, 12, 586–597.
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3, e293.
- Augustin, E.; Paw\lowska, M.; Polewska, J.; Potega, A.; Mazerska, Z. Modulation of CYP3A4 activity and induction of apoptosis, necrosis and senescence by the anti-tumour imidazoacridinone C-1311 in human hepatoma cells. Cell Biol. Int. 2013, 37, 109–120.
- Dunstan, M.S.; Barnes, J.; Humphries, M.; Whitehead, R.C.; Bryce, R.A.; Leys, D.; Stratford, I.J.; Nolan, K.A. Novel inhibitors of NRH: Quinone oxidoreductase 2 (NQO2): Crystal structures, biochemical activity, and intracellular effects of imidazoacridin-6-ones. J. Med. Chem. 2011, 54, 6597–6611.
- Fedejko-Kap, B.; Bratton, S.M.; Finel, M.; Radominska-Pandya, A.; Mazerska, Z. Role of human UDP-glucuronosyltransferases in the biotransformation of the triazoloacridinone and imidazoacridinone antitumor agents C-1305 and C-1311: Highly selective substrates for UGT1A10. Drug Metab. Dispos. 2012, 40, 1736–1743.
- Polewska, J.; Skwarska, A.; Augustin, E.; Konopa, J. DNA-damaging imidazoacridinone C-1311 induces autophagy followed by irreversible growth arrest and senescence in human lung cancer cells. J. Pharmacol. Exp. Ther. 2013, 346, 393–405.
- Potega, A.; Dabrowska, E.; Niemira, M.; Kot-Wasik, A.; Ronseaux, S.; Henderson, C.J.; Wolf, C.R.; Mazerska, Z. The imidazoacridinone antitumor drug, C-1311, is metabolized by flavin monooxygenases but not by cytochrome P450s. Drug Metab. Dispos. 2011, 39, 1423–1432.
- Skwarska, A.; Augustin, E.; Beffinger, M.; Wojtczyk, A.; Konicz, S.; Laskowska, K.; Polewska, J. Targeting of FLT3-ITD kinase contributes to high selectivity of imidazoacridinone C-1311 against FLT3-activated leukemia cells. Biochem. Pharmacol. 2015, 95, 238–252.
- Wiśniewska, A.; Niemira, M.; Jagiełło, K.; Potęga, A.; Świst, M.; Henderson, C.; Skwarska, A.; Augustin, E.; Konopa, J.; Mazerska, Z. Diminished toxicity of C-1748, 4-methyl-9-hydroxyethylamino-1-nitroacridine, compared with its demethyl analog, C-857, corresponds to its resistance to metabolism in HepG2 cells. Biochem. Pharmacol. 2012, 84, 30–42.
- Zhou, Q.; You, C.; Zheng, C.; Gu, Y.; Gu, H.; Zhang, R.; Wu, H.; Sun, B. 3-Nitroacridine derivatives arrest cell cycle at G0/G1 phase and induce apoptosis in human breast cancer cells may act as DNA-target anticancer agents. Life Sci. 2018, 206, 1–9.
- Zhou, Q.; Wu, H.; You, C.; Gao, Z.; Sun, K.; Wang, M.; Chen, F.; Sun, B. 1,3-dimethyl-6-nitroacridine derivatives induce apoptosis in human breast cancer cells by targeting DNA. Drug Dev. Ind. Pharm. 2019, 45, 212–221.
- Kalirajan, R.; Kulshrestha, V.; Sankar, S.; Jubie, S. Docking studies, synthesis, characterization of some novel oxazine substituted 9-anilinoacridine derivatives and evaluation for their antioxidant and anticancer activities as topoisomerase II inhibitors. Eur. J. Med. Chem. 2012, 56, 217–224.
- Kalirajan, R.; Sankar, S.; Jubie, S.; Gowramma, B. Molecular docking studies and in-silico ADMET screening of some novel oxazine substituted 9-anilinoacridines as topoisomerase II inhibitors. Indian J. Pharm. Educ. Res. 2017, 51, 110–115.
- Kalirajan, R.; Kulshrestha, V.; Sankar, S. Synthesis, characterization and antitumour activity of some novel oxazine substituted 9-anilinoacridines and their 3D-QSAR studies. Indian J. Pharm. Sci. 2018, 80, 921–929.
- Cheung-Ong, K.; Song, K.T.; Ma, Z.; Shabtai, D.; Lee, A.Y.; Gallo, D.; Heisler, L.E.; Brown, G.W.; Bierbach, U.; Giaever, G. Comparative chemogenomics to examine the mechanism of action of DNA-targeted platinum-acridine anticancer agents. ACS Chem. Biol. 2012, 7, 1892–1901.
- Ding, S.; Qiao, X.; Kucera, G.L.; Bierbach, U. Design of a platinum–acridine–endoxifen conjugate targeted at hormone-dependent breast cancer. Chem. Commun. 2013, 49, 2415–2417.
- Fahrenholtz, C.D.; Ding, S.; Bernish, B.W.; Wright, M.L.; Zheng, Y.; Yang, M.; Yao, X.; Donati, G.L.; Gross, M.D.; Bierbach, U. Design and cellular studies of a carbon nanotube-based delivery system for a hybrid platinum-acridine anticancer agent. J. Inorg. Biochem. 2016, 165, 170–180.
- Graham, L.A.; Suryadi, J.; West, T.K.; Kucera, G.L.; Bierbach, U. Synthesis, aqueous reactivity, and biological evaluation of carboxylic acid ester-functionalized platinum–acridine hybrid anticancer agents. J. Med. Chem. 2012, 55, 7817–7827.
- Kostrhunova, H.; Malina, J.; Pickard, A.J.; Stepankova, J.; Vojtiskova, M.; Kasparkova, J.; Muchova, T.; Rohlfing, M.L.; Bierbach, U.; Brabec, V. Replacement of a thiourea with an amidine group in a monofunctional platinum–acridine antitumor agent. Effect on DNA interactions, DNA adduct recognition and repair. Mol. Pharm. 2011, 8, 1941–1954.
- Pickard, A.J.; Liu, F.; Bartenstein, T.F.; Haines, L.G.; Levine, K.E.; Kucera, G.L.; Bierbach, U. Redesigning the DNA-Targeted Chromophore in Platinum–Acridine Anticancer Agents: A Structure–Activity Relationship Study. Chem.-Eur. J. 2014, 20, 16174–16187.
- Qiao, X.; Zeitany, A.E.; Wright, M.W.; Essader, A.S.; Levine, K.E.; Kucera, G.L.; Bierbach, U. Analysis of the DNA damage produced by a platinum–acridine antitumor agent and its effects in NCI-H460 lung cancer cells. Metallomics 2012, 4, 645–652.
- Smyre, C.L.; Saluta, G.; Kute, T.E.; Kucera, G.L.; Bierbach, U. Inhibition of DNA synthesis by a platinum–acridine hybrid agent leads to potent cell kill in nonsmall cell lung cancer. ACS Med. Chem. Lett. 2011, 2, 870–874.
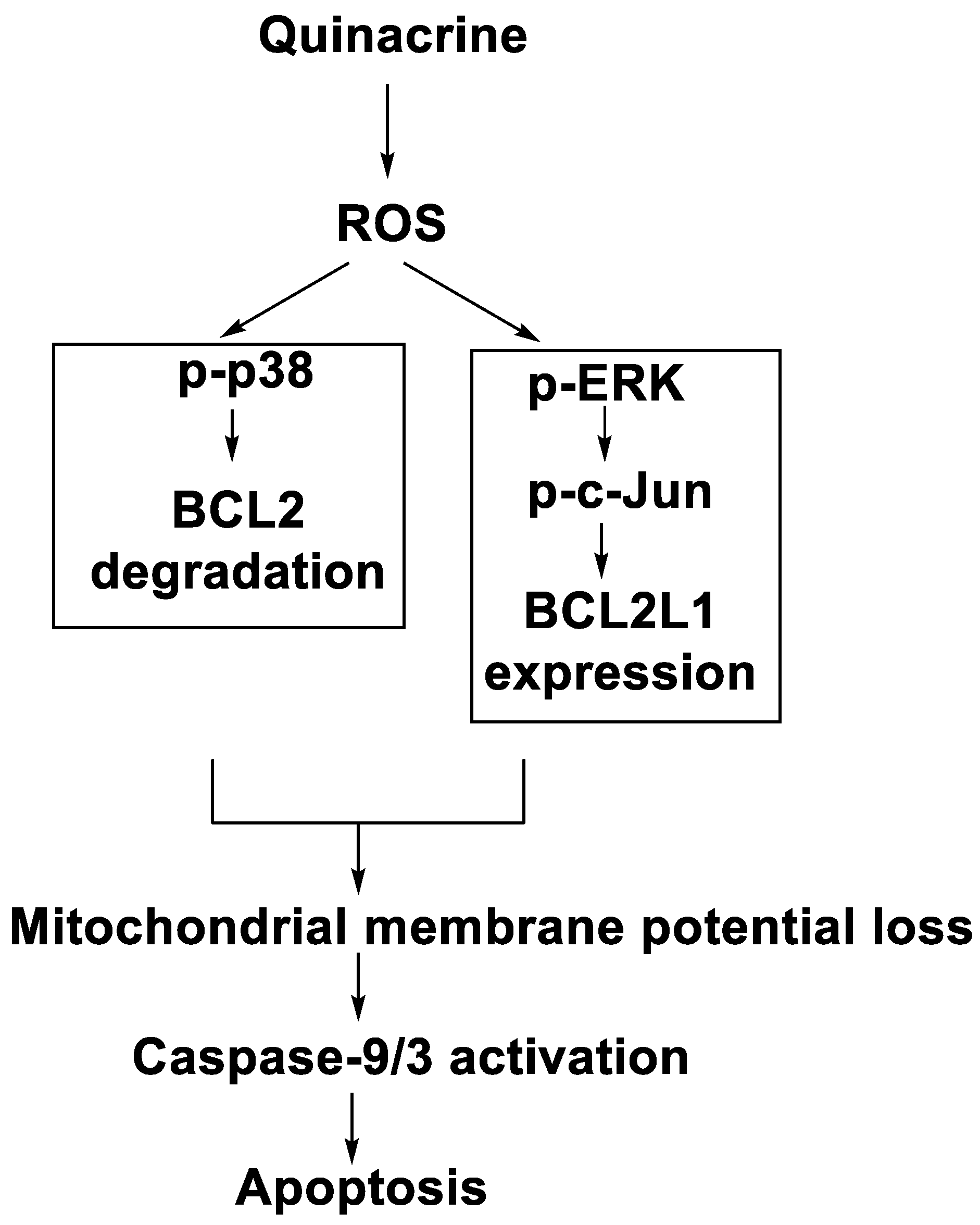
- Changchien, J.-J.; Chen, Y.-J.; Huang, C.-H.; Cheng, T.-L.; Lin, S.-R.; Chang, L.-S. Quinacrine induces apoptosis in human leukaemia K562 cells via p38 MAPK-elicited BCL2 down-regulation and suppression of ERK/c-Jun-mediated BCL2L1 expression. Toxicol. Appl. Pharmacol. 2015, 284, 33–41.
- Huang, C.-H.; Lee, Y.-C.; Chen, Y.-J.; Wang, L.-J.; Shi, Y.-J.; Chang, L.-S. Quinacrine induces the apoptosis of human leukaemia U937 cells through FOXP3/miR-183/β-TrCP/SP1 axis-mediated BAX upregulation. Toxicol. Appl. Pharmacol. 2017, 334, 35–46.
- Nayak, D.; Tripathi, N.; Kathuria, D.; Siddharth, S.; Nayak, A.; Bharatam, P.V.; Kundu, C. Quinacrine and curcumin synergistically increased the breast cancer stem cells death by inhibiting ABCG2 and modulating DNA damage repair pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105682.
- Siddharth, S.; Nayak, D.; Nayak, A.; Das, S.; Kundu, C.N. ABT-888 and quinacrine induced apoptosis in metastatic breast cancer stem cells by inhibiting base excision repair via adenomatous polyposis coli. DNA Repair 2016, 45, 44–55.
- Yang, S.; Sheng, L.; Xu, K.; Wang, Y.; Zhu, H.; Zhang, P.; Mu, Q.; Ouyang, G. Anticancer effect of quinacrine on diffuse large B-cell lymphoma via inhibition of MSI2-NUMB signaling pathway Corrigendum in/10.3892/mmr. 2020.11813. Mol. Med. Rep. 2018, 17, 522–530.
- Solomon, V.R.; Pundir, S.; Le, H.-T.; Lee, H. Design and synthesis of novel quinacrine--thiazinan-4-one hybrids for their anti-breast cancer activity. Eur. J. Med. Chem. 2017, 143, 1028–1038.
- Solomon, V.R.; Almnayan, D.; Lee, H. Design, synthesis and characterization of novel quinacrine analogs that preferentially kill cancer over non-cancer cells through the down-regulation of Bcl-2 and up-regulation of Bax and Bad. Eur. J. Med. Chem. 2017, 137, 156–166.
- Rego, M.J.d.; de Moura, R.O.; Jacob, I.T.; Lins, T.U.L.e.; Pereira, M.C.; Lima, M.d.C.A.; Galdino-Pitta, M.R.; Pitta, I.d.; Pitta, M.G.d. Synthesis and anticancer evaluation of thiazacridine derivatives reveals new selective molecules to hematopoietic neoplastic cells. Comb. Chem. High Throughput Screen. 2017, 20, 713–718.
- Barros, F.W.; Bezerra, D.P.; Ferreira, P.M.; Cavalcanti, B.C.; Silva, T.G.; Pitta, M.G.; de Lima, M.d.C.; Galdino, S.L.; Pitta, I.d.; Costa-Lotufo, L.V. Inhibition of DNA topoisomerase I activity and induction of apoptosis by thiazacridine derivatives. Toxicol. Appl. Pharmacol. 2013, 268, 37–46.
- Chagas, M.B.O.; Cordeiro, N.C.C.; Marques, K.M.R.; Pitta, M.G.R.; Rêgo, M.; Lima, M.C.A.; Pitta, M.G.R.; Pitta, I.R. New thiazacridine agents: Synthesis, physical and chemical characterization, and in vitro anticancer evaluation. Hum. Exp. Toxicol. 2017, 36, 1059–1070.
- Lafayette, E.A.; de Almeida, V.; Mônica, S.; Pitta, M.G.d.; Beltrão, E.I.C.; da Silva, T.G.; de Moura, R.O.; Pitta, I.d.; de Carvalho, L.B.; de Lima, M.D.C.A. Synthesis, DNA binding and topoisomerase I inhibition activity of thiazacridine and imidazacridine derivatives. Molecules 2013, 18, 15035–15050.
- Cui, Z.; Chen, S.; Wang, Y.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur. J. Med. Chem. 2017, 136, 372–381.
- Karelou, M.; Kourafalos, V.; Tragomalou, A.P.; Marakos, P.; Pouli, N.; Tsitsilonis, O.E.; Gikas, E.; Kostakis, I.K. Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules 2020, 25, 4584.