Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Organ microenvironments are irrefutable modules enabling the proliferation of cancer cells to organ systems exterior to the primary tumor. The hepatic tumor immune microenvironment constitutes cellular and non-cellular components including a complex mixture of immune cells.
- hepatocellular carcinoma
- tumor immune microenvironment
- immunotherapy
1. Tumor-Associated Neutrophils: New Felon in HCC Tumor Immune Microenvironment
Among other myeloid cell populations that are generated in the bone marrow, neutrophils are the most abundant leukocytes present in the circulation and are the first to be recruited at the site of infection or injury, where they act as a first line of defense. Emerging data educate that neutrophils are critical for tumor initiation, proliferation, vascularization, migration, and invasion through several mechanisms, ranging from direct support to tumor cell survival, causing immunosuppression and genetic mutations [1]. Infiltrating tumor-associated neutrophils (TANs) display anti-tumorigenic or pro-tumorigenic phenotypes. At the early stage of HCC, neutrophils are primarily located at the periphery of the tumors and exhibit anti-tumoral phenotype, whereas at the later stage, their pro-tumoral properties dominate, supporting cancer progression [2]. The pro-tumoral or anti-tumoral phenotype of neutrophils in the primary tumor or metastatic site is highly reliant on the cytokine milieu. The anti-tumorigenic neutrophils are classified as N1, while the pro-tumorigenic neutrophils are categorized as N2 phenotypes [3]. N1 neutrophils direct tumor cell cytotoxicity, constraining the development of cancer, while N2 TANs endorse cancer development through supporting carcinogenesis, angiogenesis, tumor growth, and metastasis by destroying anti-cancer immunity and synthesizing neutrophil extracellular traps (NETs) [4][5]. Establishment of NETs is initiated by the innate immune receptors through downstream intracellular mediators comprising ROS, which are generated by NADPH oxidase or mitochondria, activating myeloperoxidase, neutrophil elastase, and protein-arginine deiminase type 4 (PAD4) to promote chromatin decondensation [6][7]. NETs release occurs primarily via a cell death process called NETosis; however, live cells can also trigger the release of NETs. Systemic effects of tumors modulate NETosis, instigating NET-associated complications in cancer. Upregulation of granulocyte colony stimulating factor (G-CSF) in HCC increases systemic NETosis and transforms HCC into a high-grade phenotype [8][9].
The presence of G-CSF and tumor necrosis factor-alpha (TNF-α) in the pro-tumoral area induces the immunosuppressive profile in neutrophils by augmenting programmed death-ligand 1 (PD-L1) expression [10]. Moreover, cancer-associated fibroblasts (CAFs) infiltrating HCC stimulate the activation and survival of TANs, shown by higher expression of CD66b, PD-L1, CXCL8, TNF-α, and chemokine ligand 2 (CCL2), where enrichment of CD66b+ neutrophils in the peritumoral region is associated with decreased overall survival of patients [11]. CAFs secrete cardiotrophin-like cytokine factor 1 (CLCF1) inducing the production of CXCL6 and transforming growth factor-β (TGF-β) by tumor cells deriving TANs polarization into N2 phenotype and promoting tumor stemness [12]. CLCF1-CXCL6/TGF-β axis supports the recruitment of N2 TANs along with the regulation of cancer stemness, contributing to the poor prognosis of HCC patients. HCC-CAF-primed neutrophils suppress T cell activity by efficiently suppressing the T cell proliferation and IFN-γ production mainly through neutralization of PD-L1, which attenuated T cell suppression mediated by CAF-primed neutrophils, confirming that neutrophil mediates immune suppression through programmed death PD-1/PD-L1 pathway. In the HCC microenvironment, neutrophils act as a primary source of MMP-9 encouraging angiogenesis by the release of pro-angiogenic factors [13]. Furthermore, TANs secrete CCL2 and CCL17, which correlate with tumor size, microvascular invasion, extent of tumor differentiation, staging, and poor survival time [14].
Neutrophils in intratumoral regions express autophagy-specific protein LC3 and autophagosomes, deriving the continued production of pro-metastatic oncostatin M and MMP9, which advances the migration of cancer cells [15]. TME delays apoptosis in neutrophils with retained Mcl-1 and low levels of cleaved caspase-3. Inhibition of autophagy by an autophagy-specific inhibitor 3-methyladenine (3-MA) suppressed the production of these molecules and eliminated TME-mediated neutrophil survival, indicating a pro-tumorigenic role of neutrophil autophagy. Importantly, depletion of neutrophils effectively inhibited tumor angiogenesis and growth, suggesting a critical role of neutrophils during HCC [15].
2. Regulatory Dendritic Cells: Another Feather in the Cap of Immune Suppression during HCC
Dendritic cells (DCs) are the professional antigen presenting cells, capturing the pathogen or tumor-derived antigens, presenting them to the naïve T cells to generate effective immune response by differentiating them into effector T cells; however, these functions are generally weakened in the tumor microenvironment [16]. Due to the overwhelming growth, cancer cells alter their metabolism to uptake higher amounts of nutrients in tumor biomass and generate new tumor cells [17]. This aberrant tumor cell metabolism, in turn, induces extensive modifications in TME including tryptophan depletion, lactate and lipid accumulation, hyper glycolysis, and acidification, which minimizes the function of DCs leading to the incidence of tumor immune escape [18]. In fact, DCs present in TME block anti-tumor immunity by encouraging cancerous cell growth and dissemination, inducing genomic damage and advancing neovascularization. In most tumors, these cells preferentially acquire regulatory phenotypes known as regulatory dendritic cells (Dregs), that primarily serve tumor cells to escape immune mechanisms by expressing diverse immune regulatory molecules and producing higher levels of immunoregulatory cytokines, and have minimal aptitude to encourage T cell activation and proliferation [19]. Existing literature reveals that tumor-derived factors and tumor itself can transform conventional or myeloid dendritic cells into Dregs [20]; however, whether non-tumor cells in TME can also encourage Dregs differentiation is not clear. Dregs directly or indirectly maintain T cell unresponsiveness by limiting T cell polarization, Tregs, and MDSCs differentiation through the induction of indoleamine 2, 3 dioxygenase (IDO), IL-10, TGF-β and express COX-2, iNOS and arginase, affecting specific microenvironmental conditions in premalignant niches [19][21][22]. Production of IDO is triggered by vitamin D and prostaglandin E2 (PGE2), while its transcription is regulated by the signal transducer and activator of transcription 3 (STAT3), initiated by IL-6 from the tumor cells [23]. It has been proven that IL-6-treated DCs downregulate CD1a, CD83, CD86, HLA-DR and upregulate CD14 expression, inducing Dregs [23]. IDO degrades the essential amino acid tryptophan, required for protein synthesis, leading to the build-up of its metabolite, the kynurenine [24]. Whereas depletion of tryptophan may lead to the inhibition of T cell proliferation and accumulation of its metabolite may employ immune cell toxicity and enhance IDO production in DCs, leading to DC-induced immunosuppression and further contributing to tumor evasion [25]. The existence of CD14+ DCs was observed in peripheral blood and tumor tissues of HCC patients irrespective of tumor stages. These cells express inhibitory molecules including PD-1 and cytotoxic-lymphocyte antigen 4 (CTLA-4) on their surface and produce IL-10 in response to lipopolysaccharides (LPS) neutralization, which weakens the immune-suppressive effect of CD14+ DCs, suggesting that the immunosuppressive effect of these cells is driven by inhibitory molecules and IL-10 [26].
Additionally, tumors redirect the process of dendropoiesis (DCs generation) and further polarize these cells into a phenotype that commonly blocks the development of anti-tumor immunity [27], further backing tumor progression by allowing intra-tumoral neoangiogenesis and metastases. DCs capacity to direct the immune response is not an intrinsic feature; rather, it arises due to specific microenvironmental signals originated from TME, such as local cytokine milieu and other soluble factors. For instance, tumor-derived IL-10, TGF-β, and PGE2 can manifest DCs to attain regulatory rather than stimulatory functions, where IL-10 appeared most tolerogenic compared to other factors since it inhibited the maturation of these cells, restricted the expression of MHC class I and II and costimulatory molecules, and alleviated the production of inflammatory cytokines [28]. Studies reported that bone marrow-derived DCs cultured in the presence of IL-10 presented low levels of MHC class II, CD40, CD80, CD86, and IL-12p70 and abundantly produced IL-10, and were competent in expanding functional Tregs [29][30]. Peripheral blood and CD11c+ tumor-infiltrating myeloid cells express PD-1 in HCC in both humans and mice, which repressed IL-2 and IFN-γ as well as antigen-specific CD8 T cell proliferation under in vitro and in vivo settings, respectively; suggesting that immune surveillance against tumors is strongly regulated by PD-1 expression on DCs [31]. Given that Dregs are critical in deriving immunosuppression in HCC, new therapies selectively targeting immunosuppressive functions in DCs may assist in restoring immunostimulatory functions of DCs in TME.
3. Tumor-Associated Macrophages: One of the Gritty Culprit
Macrophages belong to the mononuclear lineage, originating and differentiating from myeloid progenitors and circulating monocytes. During the embryonic hematopoiesis, monocytes are recruited from the bone marrow reaching to different tissues and organs via traveling through circulation and then differentiating into tissue-resident macrophages, with exceptional plasticity and functional diversity. Nevertheless, one needs to bear in mind that not all the macrophages are derived from circulating monocytes including Kupffer cells (KCs) in the liver [32]. In recent years, one of the most important paradigm shifts in the macrophage biology is the origin of tissue macrophages. Earlier, it was believed that tissue macrophages are derived from circulating blood monocytes, while at present, it is clear that tissue macrophages are developed during the embryonic development, preserved separately from blood monocytes during homeostasis, and primarily self-renew from resident stem cells originated from fetal yolk sac [33]. Tissue-resident macrophages are a heterogeneous population that is inevitably required for performing tissue-specific functions and maintaining homeostasis. Fundamentally, macrophages are phagocytic in nature, retain antigen presentation capability, and provide non-specific defense by clearing foreign and harmful pathogens, cellular debris, and tumor cells, and further initiating specific-defense mechanisms [34]. Depending on the surrounding environment and cytokine milieu, macrophages can differentiate into classically activated macrophages (M1) and alternatively activated macrophages (M2), which can convert into each other owing to the changes in the internal environment. M1 macrophages are induced by T helper type 1 (Th1) cell signature cytokine IFN-γ and/or toll-like receptor (TLR) ligands, whereas M2 macrophages are encouraged through the stimulation of Th2-associated cytokines, such as IL-4 and IL-13 along with macrophage colony stimulating factor (M-CSF), TGF-β, and glucocorticoids [35]. Hepatic macrophages, referred to as KCs, can remove microbial products and other deleterious substances transported to the liver via the blood circulation. The hepatic parenchyma possesses approximately 80% of all macrophages of the body and is traversed by blood monocytes; however, this occurs only during liver injury. Normally, tissue-resident macrophages are acknowledged as M2-like phenotypes with a fundamental role in tissue homeostasis immune surveillance and resolution of inflammation by controlling excessive immune activation. Since the liver is persistently exposed to antigens from the gut and low levels of bacterial endotoxins, several mechanisms suppress incidental immune activation including M2 macrophages; therefore, the liver acts as an immunotolerant organ [36]. Tissue macrophages express pattern recognition receptors, such as toll-like receptors (TLRs), NOD-like receptors (NLRs), lectins and scavenger receptors that recognize a wide variety of PAMPs and DAMPs. Expression of heterogeneous receptors on macrophages indicates its involvement in the activation of different classes of immune response to different pathogens and viruses. Tumor-associated macrophages (TAMs) develop a pro-inflammatory microenvironment and are a potential source of steatosis-stimulated Wnt expression. The active Wnt/β-catenin signaling in macrophages may encourage the growth of tumor progenitor cells, increasing the risk of HCC as well as cholangiocarcinoma in obese individuals [37].
Tumor-associated macrophages secrete different cytokines, such as IL-6, IL-10 and TNF-α, chemokines CCL17, CCL22, CCL24, CXCL12, CXCL8 and growth factors TGF-β, vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), along with other soluble factors, such as osteopontin, cyclooxygenase-2, and matrix metalloproteinase (MMPs), modulating tissue architecture and favoring tumor cell migration, invasion, cancer progression, and metastasis [38][39][40]. TME constitutes two types of TAMs containing tissue-resident and infiltrated macrophages. During HCC, most tumor cells support the infiltration of macrophages into the tumors by displaying higher glypican-3 expression where these cells play a bigger role in HCC progression by dictating various inflammatory lesions than those of resident macrophages, which produce chemokines, deriving leukocytes influx to induce inflammation [41]. Higher TAM infiltration is frequently correlated with poor clinical outcomes in various types of tumors and reduced response to standard cancer therapeutics including chemotherapy, radiotherapy, as well as targeted therapy; therefore, limiting their infiltration could be beneficial to overcome these obstacles [42]. However, TAMs are not deleterious completely; they confer protection by eliminating the tumor. Macrophages eradicate apoptotic cells and cell debris to support the recurrence of homeostasis during the resolution of inflammation and participate in the formation of vascularized granulation tissue, epithelization, and subsidizing scar formation to every stage of damage repair [43]. However, the moral functional abilities of TAMs in TME are dominated by their ruthless behavior.
4. Myeloid-Derived Suppressor Cells: Frenemy of HCC
In the recent decade, myeloid-derived suppressor cells (MDSCs) attracted increased attention in different cancers including HCC. MDSCs are pathologically activated cells that are involved in immune regulation in different disease conditions including chronic infections, auto-immunity, sepsis, and cancer, and are strongly associated with poor clinical outcomes, especially in cancer [44]. MDSCs are classified into two different phenotypes including granulocytic/polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) conforming to their origin from granulocytic or monocytic myeloid cell lineages [45]. Nevertheless, these cells share common biological features including upregulated expression of arginase-1 (arg-1) and STAT3, and induced endoplasmic reticulum (ER) stress. Whereas, they differ in other features, such as PMN-MDSCs mediate immune suppression favorably using reactive oxygen species (ROS), arg-1, and PGE2, while M-MDCs utilize nitric oxide, immunosuppressive cytokines including IL-10 and TGF-β and immunoregulatory molecules, such as PD-L1 [45]. The main characteristics delineating MDSCs is their strong immunosuppressive nature. These cells can control immune responses mediated by diverse immune cells including T and B cells, and NK cells. MDCSs are thoroughly dispersed in the bone marrow, spleen, and peripheral blood. The presence of MDSCs has been detected in the TME where they participate in the tumor growth and angiogenesis [46]. Therefore, MDSCs is a potential approach for enhancing the current treatment of cancers. During cancer, MDSCs acquire several changes in their phenotype, functions, and metabolic processes. It is well established that metabolic reprogramming occurs during cancer, which is essential for the tumor cells to withstand their high-energy requirement to accelerate their proliferation, differentiation, and survival. MDSCs struggle for the nutrients and oxygen in the TME and thereby acclimatize their metabolism in accordance with the TME [45]. MDSCs act vigorously, selecting the most competent metabolic pathways to endure their regulatory, suppressive, and pro-tumorigenic functions. Lipid, glucose, and amino acid metabolism are altered in MDSCs where lipid metabolism is crucial for its differentiation and functions, while glucose metabolism supports their maturation from bone marrow precursors [47][48]. In addition, upregulated glycolytic pathways defend MDSCs from apoptosis and participate in their survival by preventing ROS-mediated apoptosis. Under hypoxic conditions, the activation of hypoxia-inducible factor-1α (HIF-1α) triggers high glycolysis in MDSCs [49]. Overexpression of HIF-1α is highly associated in supporting tumor growth and metastasis by instigating angiogenesis and regulating cellular metabolism to overcome hypoxia. In the case of MDSCs, HIF-1α regulates the differentiation and function of MDSCs in the TME and, in fact, facilitates the differentiation of M-MDCs into tumor-associated macrophages [49].
The frequencies of circulating MDSCs are increased during HCC and directly correlate with the combative tumor features and volume [50]. Tumor-infiltrating leukocytes consisted of the highest percentage of MDSCs expressing PD-L1 than those present in the liver-infiltrating leukocytes and peripheral blood. Several lines of evidence pointed toward prognostic implications and translational significance of MDSCs in HCC. A study by Nan et al. identified a novel marker, LOX-1 (lectin-like oxidized low-density lipoprotein (LDL) receptor-1) in PMN-MDSCs in HCC patients and found that LOX-1+CD15+ cells were increased in the peripheral blood and their frequencies were associated with those present in HCC tissue [51]. These cells inhibited the proliferation of IFN-γ producing T cells under in vitro setting, while their LOX-1-CD15+ counterpart did not; confirming the role of LOX-1 in MDSC-derived immune suppression. MDSCs influence the tumor microenvironment by producing several angiogenic factors and vascular-modulating enzymes, such as bombina variegata peptide 8 (Bv8, a homolog of endocrine gland-derived vascular endothelial growth factor) through G-CSF dependent STAT3 signaling, promoting angiogenesis and hematopoietic cell mobilization. Additionally, MDSCs could obtain endothelial cell properties in TME, directly incorporating into tumor endothelium [52]. Reversing the pro-tumor effects of MDSCs could be attained by their depletion, inhibiting their trafficking and migration into TME, and restricting their immunosuppressive functions.
5. Regulatory T Cells: Persistent Suppressors of Anti-Tumor Immunity
Regulatory T cells (Tregs) are indispensable for maintaining peripheral tolerance by suppressing the immune response, which confines autoimmune diseases and chronic inflammatory diseases. However, they are also involved in the development and progression of tumors by restricting effective anti-tumor immunity, and are linked to poor survival in different cancers, serving as a critical target to explore [53]. The existence of two types of Tregs has been proven: (1) Natural Tregs (nTregs) and (2) Induced Tregs (iTregs) [54]. Naturally occurring Tregs develop as a distinct lineage in the thymus and specifically express forkhead box P3 (FOXP3) transcription factor in their nucleus and CD25 and CTLA-4 on their surface, maintaining immunological tolerance and homeostasis, while induced Tregs develop from naïve conventional T cells in the periphery after receiving an antigenic signal [54]. Few studies indicate that iTregs are CD4+ and FOXP3, whereas others suggest that both Tregs unanimously express FOXP3 and participate in immunological tolerance in circumstances where nTregs are reduced or functionally defective [55]. A significant amount of effort has been made to uncover the mechanisms of Tregs’ suppressive activity in tumor immunity. Generally, Tregs modify the effector cell function at different stages of the immune response. At an early stage, Tregs limit T cell activation and proliferation by the induction of genes involved in growth arrest or inhibition of cell proliferation, while at the later stage, they control the differentiation and function of effector cells by producing suppressive cytokines, inhibiting effector cell migration, and causing metabolic disruption [56].
Tregs target a wide range of immune cells, such as dendritic cells, macrophages, NK cells and neutrophils, CD4+, CD8+ T cells and B cells, and are suspected to have lysing effect on these cells. Chemokine ligands CCL17 and CCL22 present in TME facilitate the infiltration and accumulation of Tregs in tumor tissue through chemokine receptor 4 (CCR4), a G protein-coupled receptor, primarily expressed on the most immunosuppressive Tregs population. Recently, it has been reported that tumor-infiltrating Tregs (TIL-Tregs) are predominantly CCR4+ and are functionally more immunosuppressive than those of CCR4-Tregs [57]. Nearly 90% of TIL-Tregs are CCR4+, while peritumoral tissue and peripheral Tregs contain a significantly lower proportion of CCR4+ Tregs. Single cell gene expression database of tumor infiltrating Tregs (TIL-Tregs) exposed that these cells have higher expression of genes involved in Tregs proliferation and exhaustion, such as UHRF1, ID2, and CXCL13 [58], while CCR4-Tregs express granzymeH, LYN, CXCL9, and pro-melanin-concentrating hormone genes, which are involved in T cell differentiation and activation [59]. Adaptive immune resistance to immunotherapies is partly attributed to the upregulation of CCR4 ligands in the tumor, initiating increased Tregs migration to the TME. Checkpoint inhibitors, such as anti-PD-1 and anti-CTLA-4 antibodies, have provided exceptional anti-tumor responses and extended the survival of patients in several types of cancers; nevertheless, few of them fail to respond or if they do respond, they relapse later by developing resistance to immunotherapy, which is attributed to the accumulation of Tregs in the tumors [60][61]. Therefore, CCR4 antagonism is a potential approach to control Tregs accumulation in the TME. Indeed, CCR4 antagonism in combination with sorafenib showed enhanced anti-tumor efficacy by transforming the immune landscape toward anti-tumor immunity through overall reduction in Tregs, diminishing proportions of CCR4+ TIL-Tregs, decreasing PD-1+CD8+ T cells, and enhancing IFN-γ+CD8+ T cells [57]. Additionally, CCR4 antagonist reduced the tumor growth of CCL17high and CCL22high tumors, suggesting that CCR4 blockade is beneficial not only for controlling Tregs infiltration and reversing CD8 T cell exhaustion, but also for restricting tumor growth which can be particularly beneficial in those who develop sorafenib resistance. One of the main challenges of targeting tumor Tregs is to specifically reduce tumor Tregs without affecting the normal tissue and peripheral pools where they are still needed to perform their normal function and maintain immune tolerance. CCR4 inhibition with CCR4-351 is a promising target in that aspect as it appears to be very selective in averting Tregs migration into TME without affecting the other pools of Tregs present in peripheral blood, skin, and spleen. This finding most likely can be translated into humans where the tumor has higher CCL17/CCL22 levels since depletion of systemic Tregs has the risk of inducing autoimmunity [62]. Overall, the function of immune cells in HCC has been summarized in Figure 1.
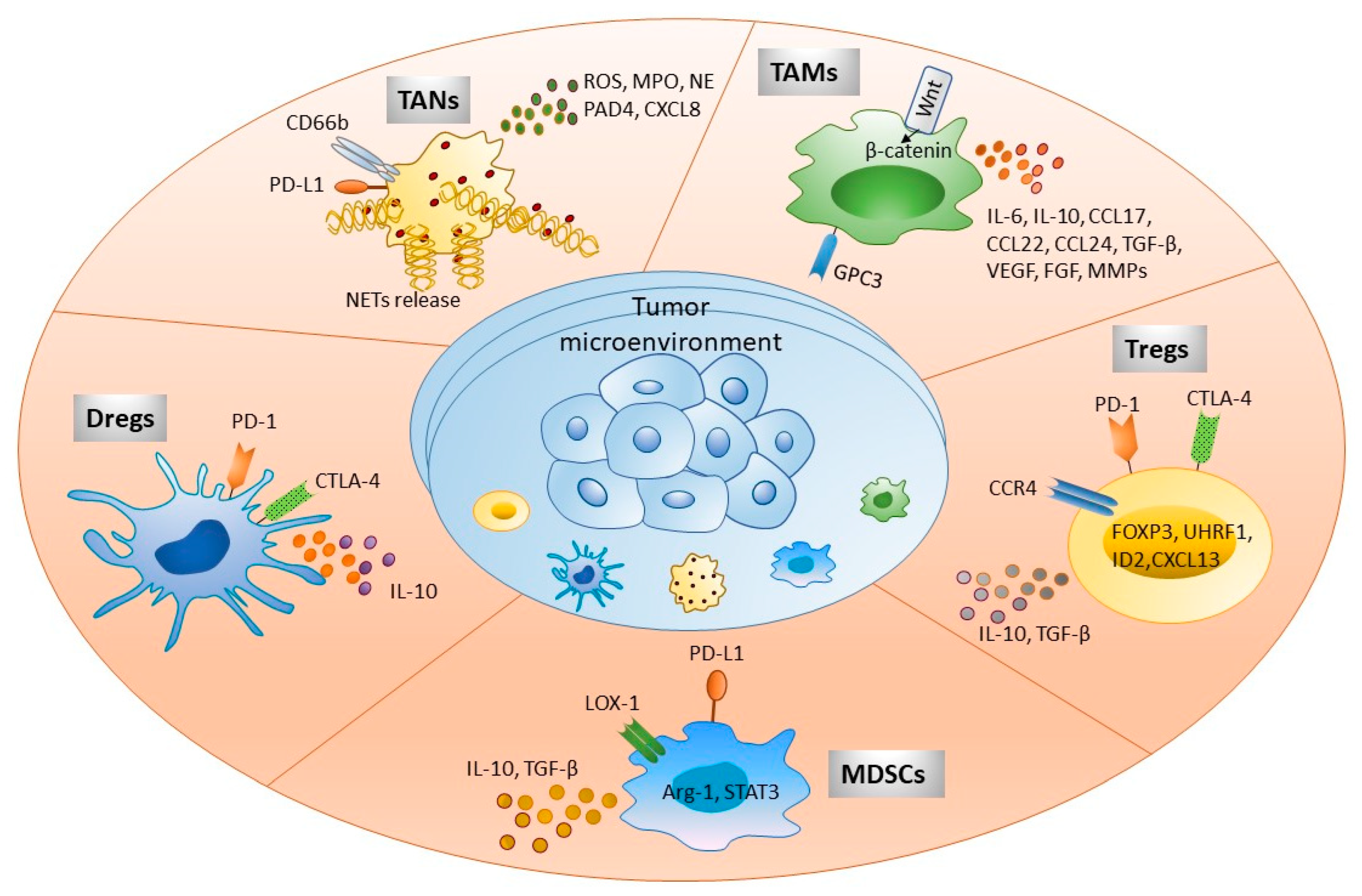
Figure 1. Immune cells present in tumor microenvironment participate in HCC development. Tumor immune microenvironment exhibits various immune cells as shown in the figure. These cellular populations interact with other parenchymal, nonparenchymal, and tumor cells directly by expressing different cell surface molecules or via secretory factors, such as cytokines, chemokines, and other soluble factors. This complex cellular interplay significantly participates in immune escape, tumor invasion, and metastasis. Targeting these molecules serves as a potential immunotherapeutic strategy to treat HCC patients. Abbreviations: FOXP3, Forkhead box protein 3; ROS, Reactive oxygen species; MPO, Myeloperoxidase; NE, Neutrophil elastase; PAD4, Protein-arginine deiminase type 4; NETs, Neutrophil extracellular traps; GPC3, Glypican 3; ILs, Interleukin; CCL and CXCL, Chemokine ligand; TGF-β, Transforming growth factor-β; VEGF, Vascular endothelial growth factor; FGF, Fibroblast growth factor; MMPs, Matrix metalloproteinases; CTLA-4, Cytotoxic T lymphocyte antigen 4; CCR4, Chemokine receptor 4; LOX-1, Lectin-like oxidized low-density lipoprotein (LDL) receptor-1.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24010437
References
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503.
- Mackey, J.B.G.; Coffelt, S.B.; Carlin, L.M. Neutrophil Maturity in Cancer. Front. Immunol. 2019, 10, 1912.
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955.
- Gregory, A.D.; Houghton, A.M. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011, 71, 2411–2416.
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081.
- Chen, T.; Li, Y.; Sun, R.; Hu, H.; Liu, Y.; Herrmann, M.; Zhao, Y.; Muñoz, L.E. Receptor-Mediated NETosis on Neutrophils. Front. Immunol. 2021, 12, 775267.
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360.
- Van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360.
- Yang, L.Y.; Luo, Q.; Lu, L.; Zhu, W.W.; Sun, H.T.; Wei, R.; Lin, Z.F.; Wang, X.Y.; Wang, C.Q.; Lu, M.; et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J. Hematol. Oncol. 2020, 13, 3.
- Sun, R.; Xiong, Y.; Liu, H.; Gao, C.; Su, L.; Weng, J.; Yuan, X.; Zhang, D.; Feng, J. Tumor-associated neutrophils suppress antitumor immunity of NK cells through the PD-L1/PD-1 axis. Transl. Oncol. 2020, 13, 100825.
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422.
- Song, M.; He, J.; Pan, Q.Z.; Yang, J.; Zhao, J.; Zhang, Y.J.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology 2021, 73, 1717–1735.
- Kuang, D.M.; Zhao, Q.; Wu, Y.; Peng, C.; Wang, J.; Xu, Z.; Yin, X.Y.; Zheng, L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J. Hepatol. 2011, 54, 948–955.
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17.
- Li, X.F.; Chen, D.P.; Ouyang, F.Z.; Chen, M.M.; Wu, Y.; Kuang, D.M.; Zheng, L. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J. Hepatol. 2015, 62, 131–139.
- Ormandy, L.A.; Farber, A.; Cantz, T.; Petrykowska, S.; Wedemeyer, H.; Horning, M.; Lehner, F.; Manns, M.P.; Korangy, F.; Greten, T.F. Direct ex vivo analysis of dendritic cells in patients with hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 3275–3282.
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47.
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154.
- Ma, Y.; Shurin, G.V.; Gutkin, D.W.; Shurin, M.R. Tumor associated regulatory dendritic cells. Semin. Cancer Biol. 2012, 22, 298–306.
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44.
- Shurin, M.R.; Naiditch, H.; Zhong, H.; Shurin, G.V. Regulatory dendritic cells: New targets for cancer immunotherapy. Cancer Biol. Ther. 2011, 11, 988–992.
- Liu, Q.; Zhang, C.; Sun, A.; Zheng, Y.; Wang, L.; Cao, X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J. Immunol. 2009, 182, 6207–6216.
- Cheng, J.T.; Deng, Y.N.; Yi, H.M.; Wang, G.Y.; Fu, B.S.; Chen, W.J.; Liu, W.; Tai, Y.; Peng, Y.W.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198.
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248.
- Van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34.
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2014, 59, 567–579.
- Enk, A.H.; Jonuleit, H.; Saloga, J.; Knop, J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int. J. Cancer 1997, 73, 309–316.
- Popov, A.; Schultze, J.L. IDO-expressing regulatory dendritic cells in cancer and chronic infection. J. Mol. Med. 2008, 86, 145–160.
- Kalantari, T.; Ciric, B.; Kamali-Sarvestani, E.; Rostami, A. Bone marrow dendritic cells deficient for CD40 and IL-23p19 are tolerogenic in vitro. Iran. J. Basic Med. Sci. 2020, 23, 287–292.
- Marguti, I.; Yamamoto, G.L.; da Costa, T.B.; Rizzo, L.V.; de Moraes, L.V. Expansion of CD4+ CD25+ Foxp3+ T cells by bone marrow-derived dendritic cells. Immunology 2009, 127, 50–61.
- Lim, T.S.; Chew, V.; Sieow, J.L.; Goh, S.; Yeong, J.P.; Soon, A.L.; Ricciardi-Castagnoli, P. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology 2016, 5, e1085146.
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56.
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35.
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737.
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13.
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2019, 10, 3112.
- Wang, K.; Qiu, X.; Zhao, Y.; Wang, H.; Chen, L. The Wnt/β-catenin signaling pathway in the tumor microenvironment of hepatocellular carcinoma. Cancer Biol. Med. 2021, 19, 305–318.
- Wan, S.; Kuo, N.; Kryczek, I.; Zou, W.; Welling, T.H. Myeloid cells in hepatocellular carcinoma. Hepatology 2015, 62, 1304–1312.
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248.
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. BioMed Res. Int. 2013, 2013, 187204.
- Takai, H.; Ashihara, M.; Ishiguro, T.; Terashima, H.; Watanabe, T.; Kato, A.; Suzuki, M. Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer Biol. Ther. 2009, 8, 2329–2338.
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226.
- Gordon, S.; Plüddemann, A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front. Immunol. 2018, 9, 127.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498.
- Ostrand-Rosenberg, S.; Beury, D.W.; Parker, K.H.; Horn, L.A. Survival of the fittest: How myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol. Immunother. 2020, 69, 215–221.
- Yan, D.; Yang, Q.; Shi, M.; Zhong, L.; Wu, C.; Meng, T.; Yin, H.; Zhou, J. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur. J. Immunol. 2013, 43, 2943–2955.
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119.
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365.
- Ma, C.; Zhang, Q.; Greten, T.F. MDSCs in liver cancer: A critical tumor-promoting player and a potential therapeutic target. Cell. Immunol. 2021, 361, 104295.
- Nan, J.; Xing, Y.F.; Hu, B.; Tang, J.X.; Dong, H.M.; He, Y.M.; Ruan, D.Y.; Ye, Q.J.; Cai, J.R.; Ma, X.K.; et al. Endoplasmic reticulum stress induced LOX-1(+) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology 2018, 154, 144–155.
- Qu, X.; Zhuang, G.; Yu, L.; Meng, G.; Ferrara, N. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: Key role of Stat3 signaling. J. Biol. Chem. 2012, 287, 19574–19584.
- Zhao, H.Q.; Li, W.M.; Lu, Z.Q.; Yao, Y.M. Roles of Tregs in development of hepatocellular carcinoma: A meta-analysis. World J. Gastroenterol. 2014, 20, 7971–7978.
- Workman, C.J.; Szymczak-Workman, A.L.; Collison, L.W.; Pillai, M.R.; Vignali, D.A. The development and function of regulatory T cells. Cell. Mol. Life Sci. 2009, 66, 2603–2622.
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711.
- Sukiennicki, T.L.; Fowell, D.J. Distinct molecular program imposed on CD4+ T cell targets by CD4+CD25+ regulatory T cells. J. Immunol. 2006, 177, 6952–6961.
- Gao, Y.; You, M.; Fu, J.; Tian, M.; Zhong, X.; Du, C.; Hong, Z.; Zhu, Z.; Liu, J.; Markowitz, G.J.; et al. Intratumoral stem-like CCR4+ regulatory T cells orchestrate the immunosuppressive microenvironment in HCC associated with hepatitis B. J. Hepatol. 2022, 76, 148–159.
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16.
- Weng, N.P.; Araki, Y.; Subedi, K. The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 2012, 12, 306–315.
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86.
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N.; et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal. 2022, 20, 44.
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors establish resistance to immunotherapy by regulating T(reg) recruitment via CCR4. J. Immunother. Cancer 2020, 8, e000764.
This entry is offline, you can click here to edit this entry!