Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Heart failure (HF) is an extremely complex, multifactorial, progressive clinical condition, characterized by cardiac function impairment secondary to a structural and/or functional heart abnormality. Sodium-glucose cotransporter (SGLT2i) was the first class of glucose-lowering drugs able to reduce cardiovascular events in diabetic and non-diabetic patients with HF.
- heart failure
- pharmacological therapy
- gliflozins
- SGLT-2 inhibitors
1. Introduction
HF is an extremely complex, multifactorial, progressive clinical condition, characterized by cardiac function impairment secondary to a structural and/or functional heart abnormality [1]. The natural history of HF is characterized by progressive worsening and deterioration of patient performance status [1][2][3][4][5][6][7][8]. A total of 64.3 million people worldwide are estimated to have HF, with a prevalence of 2% in the general population and up to 12% in people older than 65 years old [4][8]. Up to 5% of all hospital admissions are due to HF, and this number is projected to increase by 50% in the next 25 years with a 30-day re-admission rate of 20–30% [4][8][9]. Ischemic heart disease remains the leading cause of HF in western countries, followed by hypertension, non-ischemic dilatative cardiomyopathy, valvular and congenital heart disease, arrhythmias and degenerative cardiomyopathies [1][10][11][12][13]. Neurohormonal and biochemical dysregulation are main actors in HF progressive worsening and performance status deterioration [1][14][15][16][17]. Other factors sucas endothelial dysfunction, chronic inflammation, oxidative stress and metabolic dysregulation have a critical role in sustaining this condition [1][4][5][6][7][18][19][20][21].
HF has been divided into distinct phenotypes based on the assessment of left ventricular (LV) systolic function, expressed through LV ejection fraction (EF) (LVEF). According to the 2021 European Society of Cardiology (ESC) guidelines for diagnosis and treatment of HF [1], researchers distinguish: (1) HFrEF in patients with reduced LVEF (LVEF ≤ 40%); (2) HFmrEF in patients with a LVEF between 41% and 49% (mildly reduced LV systolic function); and (3) HFpEF in patients with symptoms and signs of HF, with structural and/or functional cardiac abnormalities and/or raised natriuretic peptides, with an LVEF ≥ 50%.
HFrEF is characterized by a substantial cardiomyocyte loss, resulting in the development of systolic dysfunction. In HFrEF patients, the volume overload is most often the result of permanent neurohumoral activation (RAA-System).
HFpEF is characterized by structural and cellular alterations leading to an inability of the LV to relax properly, e.g., cardiomyocyte hypertrophy, intercellular fibrosis, altered cardiomyocyte relaxation and inflammation.
Differences in the pathological development of HFrEF and HFpEF have been reported for aspects of inflammation, endothelial function, cardiomyocyte hypertrophy and death and fibrosis [22].
Unlike HFrEF, the HFpEF syndrome is characterized by a systemic proinflammatory state induced by comorbidities (including obesity, diabetes mellitus (DM), chronic obstructive pulmonary disease, renal failure and hypertension) as the main cause of myocardial structural and functional alterations [23]. Systemic inflammation also affects other organs such as the lungs, skeletal muscle, and kidneys, leading, respectively, to pulmonary hypertension, muscle weakness, and sodium retention [24].
Sodium glucose cotransporter type 2 inhibitors (SGLT2-i) represent a new class of anti-hyperglycemic agents for type-2 DM (T2DM), which act insulin-independently to selectively inhibit renal glucose reabsorption, thereby increasing urinary glucose excretion [25]. SGLT2i was the first class of glucose-lowering drugs able to reduce cardiovascular events in diabetic and non-diabetic patients with HF.
2. Mechanism of Action and Effects of SGLT2-i
The mechanisms by which gliflozines impact mortality and morbidity are not completely understood, particularly on the basis that beneficial cardiovascular effects have been observed irrespective of renal function in patients with reduced glucose filtration and thus glucose excretion [26][27]. Nowadays, the most persuasive and fascinating hypothesis seems to be that gliflozins intervene on different HF models by modifying loading conditions, neurohormonal axes and heart cells’ biochemical and vascular functions, resulting in an improvement of all HF aspects. Below the effects of SGLT2i on these models will be summarized.
2.1. Cardio-Renal Model and Modifying Loading Conditions
Considering the critical role of cardiac-induced renal dysfunction in the pathophysiology of HF, it is possible to explain that the observed cardiac benefits may be embedded in modulated renocardiac signaling. Although gliflozins act mostly as diuretics, they, contrary to loop diuretics, have lasting beneficial effects on functional class, rates of HF hospitalization and mortality in patients with HF. Osmotic diuresis and natriuresis induced by SGLT-2i may help maintain the correct load condition, resulting in an improvement in blood volume [28][29]. However, this natriuresis is not associated with neurohormonal activation, potassium loss or renal dysfunction. This favorable diuretic profile provides an important advantage in the management of volume status in patients with HF and may justify the superior long-term HF outcomes. Analysis from the EMPA-REG OUTCOME trial highlighted how change in load condition was the first determinant of CV death reduction in patients treated with empagliflozin [29].
The SGLT2-i nephroprotective effect is also demonstrated by the reduction of albuminuria and macroalbuminuria; these could be linked to increased sodium delivery at the macula densa and afterwards activation of tubuloglomerular feedback, which enhances afferent arteriolar tone and decreases intraglomerular pressure [30].
Moreover, gliflozins seem to also inhibit the sodium-hydrogen exchanger on tubular cells (Figure 1), determining lower sodium and water reuptake [31]. The renal protective action of gliflozins may contribute more to the clinical benefits of gliflozins than their actions on metabolism mitochondria and vascular smooth muscle.
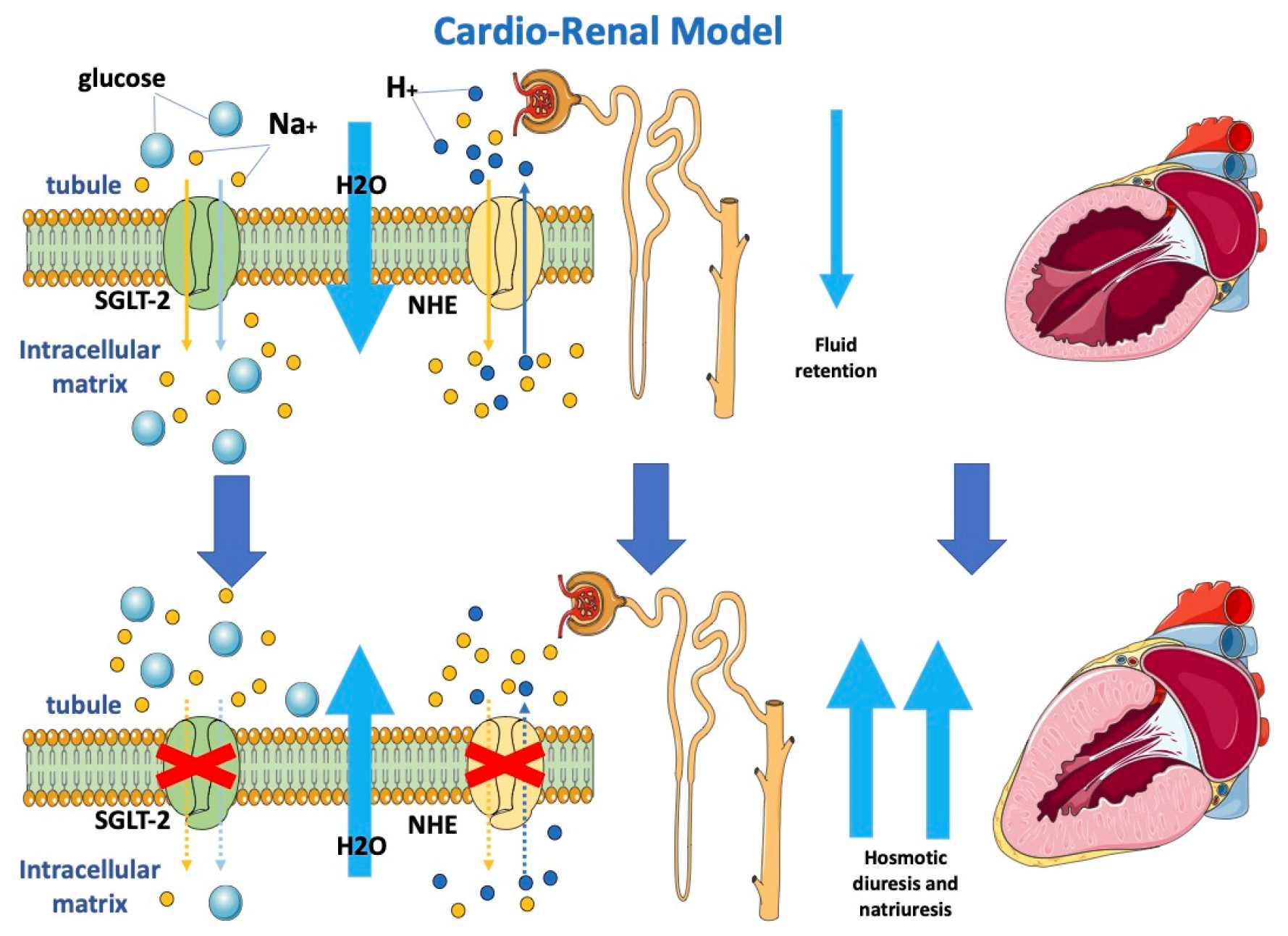
Figure 1. Effects of SGLT2-i on the cardio-renal model.
Gliflozins determine osmotic diuresis and natriuresis by inhibiting SGLT-2 and NHE, respectively, on tubular cells. Blocking both SGLT-2 and NHE determines a reduction in sodium re-uptake and, consequently, increased water loss with an improvement in loading conditions.
SGLT-2: sodium-glucose cotransporter 2; NHE: sodium-hydrogen exchanger.
2.2. Neurohormonal Model
The sympathetic nervous system (SNS) regulates glucose metabolism in various organs, including the kidneys. An important crosstalk between the SNS and SGLT2 regulation may not only explain SNS-induced modifications of glucose metabolism but may contribute to the cardiovascular and renal function improvement shown in subjects in therapy with SGLT2-i [32]. In fact, sympathetic inhibition by SGLT-2i can be regulated by the central autonomic system, and this mechanism may show how SGLT-2i improves left ventricle function [33]. Furthermore, beneficial effects of SGLT2-i are directly or indirectly related to a hemodynamic effect due to SGLT2 influence on the RAA system, including the attenuation of intracardiac fibrosis (Figure 2) [34]. A lack of increase in heart rate despite reductions in blood pressure and plasma volume may suggest decreased SNS activity [35]. Compared with classical diuretics, SGLT2-i may have different effects on the interstitial and intravascular compartments; dapagliflozin administration provides greater electrolyte-free water clearance and greater fluid clearance from the interstitial fluid space than from the circulation via peripheral sequestration of osmotically inactive sodium [26]. This may also limit the deleterious effects of reflex neurohumoral stimulation that usually occurs in response to intravascular volume depletion with traditional diuretics [36]. In HF, myocardial remodeling occurs through different pathways, resulting in apoptosis, fibroblast proliferation and fibrosis as a consequence of RAAS overactivation, hyperadrenergic tone and a chronic inflammatory state [37][38]. Sodium-proton exchanger (NHE), a transport protein, is hyperactivated in HF models [39]. NHE would determine increased cytoplasmatic levels of sodium and, consequently, calcium. High calcium cytoplasmatic levels are strongly related to myocardial cell damage and apoptosis. [38] It has been hypothesized that SGLT2i would be able to directly block NHE with a reduction in Na and fluid retention and improve cardiac remodeling through cardiac NHE inhibition and consequent intracellular calcium reduction in favor of mitochondrial calcium [40][41].
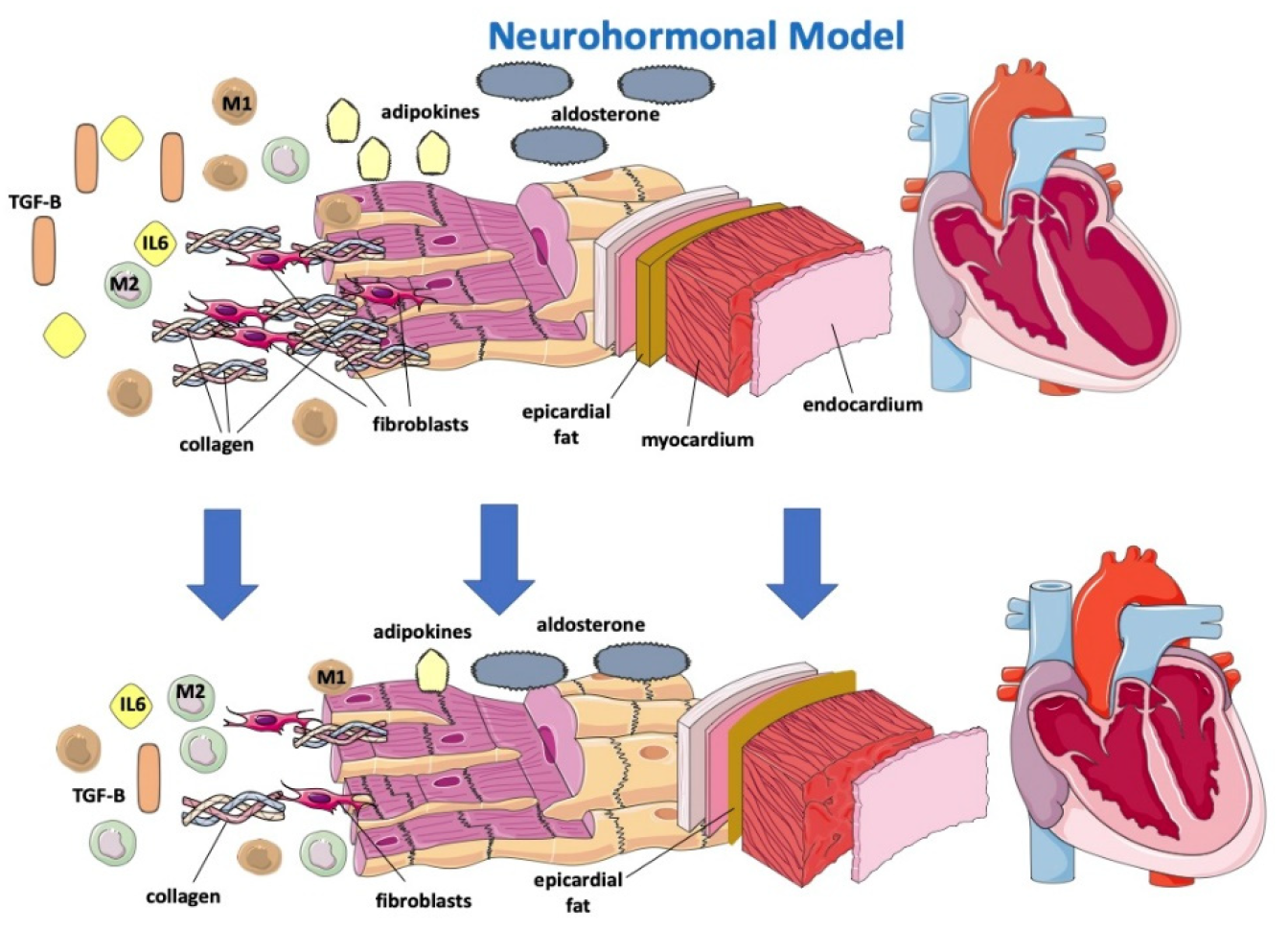
Figure 2. Effects of SGLT2-i on a neurohormonal model and on inflammatory markers.
2.3. Anti-Inflammatory Model
Gliflozins would be further effective on cardiac remodeling because of their anti-inflammatory effects [26][37][42]. In fact, SGLT2i has been proven to reduce circulating inflammatory and pro-fibrotic mediators, particularly IL-6, TGFBeta 1, which is involved in myofibroblast proliferation, and adipokines, and to reduce oxidative stress, allowing a reassertion of the M1/M2 macrophage ratio (Figure 2) [38][42][43]. SGLT2 was demonstrated to promote M2 macrophage activation through oxidative stress reduction and to determine TGF-Beta1 attenuation, resulting in a slowing of fibrous tissue deposition and cardiac remodeling processes [37][42][44][45][46].
SGLT2-i, by reducing levels of leptin (a pro-inflammatory adipokine that appears to have a considerable role in HFpEF onset), would indirectly lower aldosterone levels and, consequently, sodium reuptake and myocardial fibrosis [47]. How SGLT2i lowers leptin levels remains to be understood, and it is not simply justifiable by their effect on adipose tissue reduction [47].
Empaglifozin was associated with a reduction in epicardial fat at 6 months [48]. Epicardial fat may be an important determinant in cardiac dysfunction in light of its strict interaction with the heart. In fact, epicardial adipose tissue (EAT) is a local source of catecholamines and proinflammatory cytokines, leading macrophages to switch to the M1 phenotype [48], and thus predisposes to arrhythmias, progressive cardiomyocyte death, tissue inflammation and cardiac fibrosis and remodeling, differently from pericardial fat, which is relatively more metabolically “inert” [49].
Dapagliflozin is associated with a reduction in both atrial flutter/atrial fibrillation and ventricular arrhythmias, probably through a reduction in EAT [50]. However, reducing EAT may not be beneficial when associated with increased inflammation.
Results derived from the EMPA-TROPISM study [48] showed that empagliflozin significantly improved adiposity, interstitial myocardial fibrosis, aortic stiffness and inflammatory markers in nondiabetic patients with HFrEF. SGLT2i may be anti-inflammatory agents by acting either indirectly by improving metabolism and reducing oxidative stress or by direct modulation of inflammatory signaling pathways [51].
Gliflozins’ beneficial effect on myocardial remodeling. SGLT-2i determines the reduction of IL 6, TGF-Beta, adipokines and aldosterone, promotes reassessment of the M1/M2 macrophage ratio and reduces epicardial fat, resulting in lower apoptosis, oxidative stress, collagen deposition and fibroblast proliferation.
SGLT-2: sodium-glucose cotransporter; TGF-B: transforming growth factor—Beta; M1: M1 macrophage; M2: M2 macrophage; IL6: interleukin-6.
2.4. Cardio-Circulatory Model
From the cardio-circulatory model’s point of view, SGLT-2i would be effective in reducing afterload by reducing cardiovascular resistances (Figure 3). NHE hyperactivity leads to higher cytoplasmatic calcium levels with vascular constriction. Lowering of cytoplasmatic calcium at this level would result in vascular relaxation and, consequently, to the drop in arterial stiffness described in previous studies [40][52][53][54]. A possible interaction of dapagliflozin with the K+ voltage channel, a major determinant for resting membrane potential and thus vascular tone, would determine vasodilation [55]. Dapagliflozin was observed to directly determine increased PKG activity, another important factor involved in vasodilation [55]. The impairment in vascular function contributes to the progression of CHF by increasing the afterload. An improvement of vascular function after treatment with empagliflozin was demonstrated in HF patients [56]. Empagliflozin causes a decrease in the stiffness of the aorta and the proximal branches, reducing the afterload of the left ventricle. The decreased afterload of the LV may contribute to the beneficial effects of SGLT2 inhibition. The resulting improvement of ventricular-arterial coupling may contribute to the improved prognosis observed in patients with HF under SGLT2 inhibition.
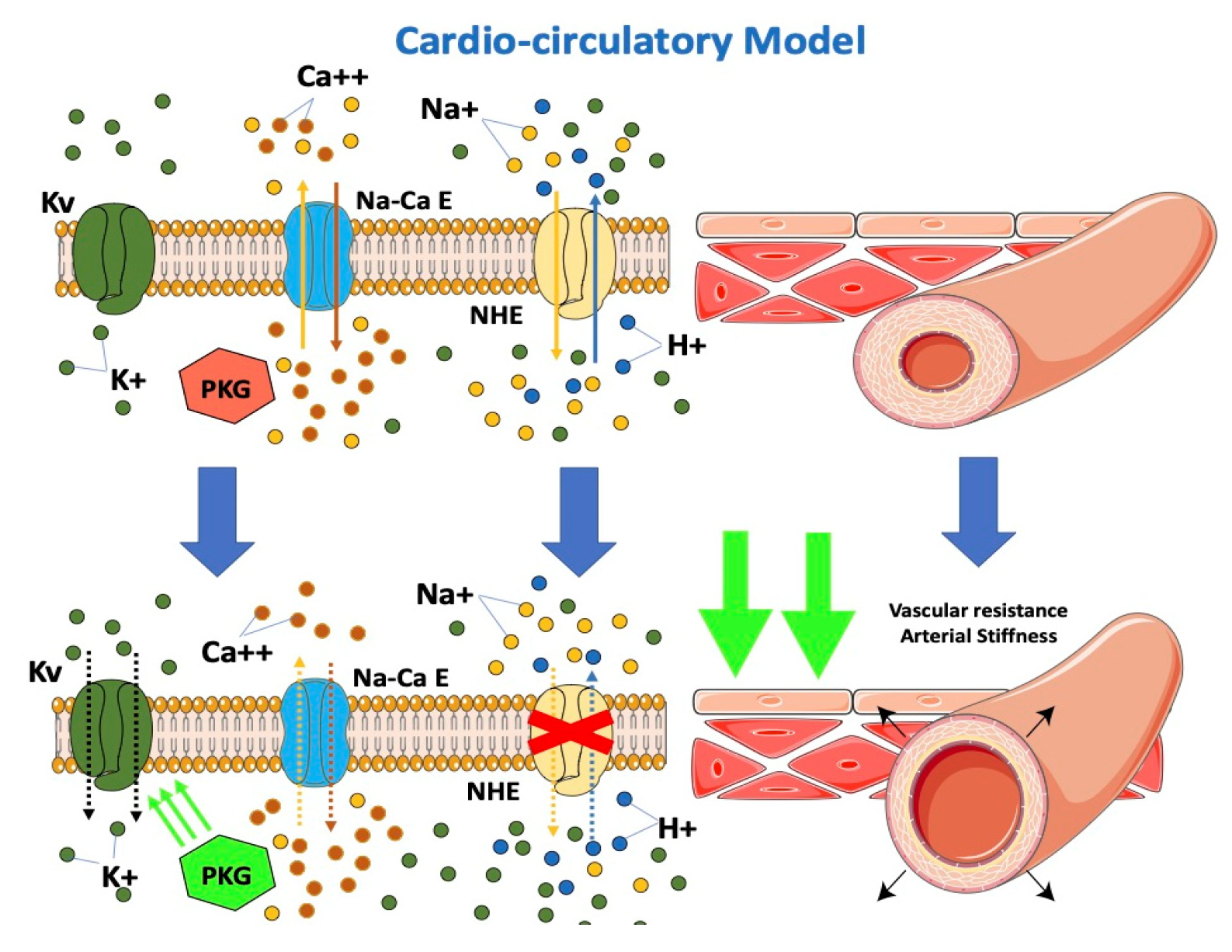
Figure 3. Effects of SGLT2-i on the cardio-circulatory model.
In an observational, non-randomized study in patients with CHF and T2DM, switching from other oral hypoglycemic drugs to SGLT2i demonstrated improved endothelial function [57]. The direct effects of SGLT2 inhibition on vascular function, combined with the natriuresis effects of SGLT2 inhibition, may contribute to hemodynamic effects [58].
Gliflozins reduce afterload by reducing cardiovascular resistance. SGLT-2i inhibits NHE expressed on endothelial and vascular smooth muscle cells, resulting in a decrease in cytoplasmatic calcium with vascular relaxation and the consequent drop in arterial stiffness. Furthermore, SGLT-2i interacts with the K+ voltage channel on vascular smooth muscle and increases PKG activity, which is involved in vascular tone.
SGLT-2: sodium-glucose cotransporter; PKG: phosphokinase: NHE: sodium-hydrogen exchanger. Kv: K+ voltage channel; Na-Ca E: Na-Ca exchanger.
2.5. Biochemical Model
In HF models, a change in the energetic metabolism of the heart has been observed with an increased use of ketones (in particular beta-hydroxybutyrate), which apparently are more oxygen-sparing [59][60]. Ketone bodies supply energy to tissues when glucose is not readily available (e.g., hypoglycemia or insulin resistance) or in the case of exceeding levels of fatty acids, such as in the case of lipolysis activation. In end-stage HF, hyperadrenergic tone determines insulin resistance and increased lipolysis with higher levels of circulating ketones [60].
A switch in the energetic substrate would be an adaptive response of a failing heart to increase its own efficiency. B-hydroxybutyrate is thought to have an oxidative stress scavenging action and to reduce oxidative stress [61]. Moreover, B-hydroxybutyrate would inhibit histone deacetylases in myocytes (Figure 4), suppressing transcription of genes involved in heart hypertrophy and fibrosis [61][62]. According to this fascinating “superfuel” theory, it would be reasonable to suppose that SGLT2i, through a not yet well understood mechanism that involves insulin level reduction and lipolysis, would improve heart efficiency and remodeling by increasing B-hydroxybutyrate levels [59][60][61][63]. This hypothesis would also, in part, possibly explain the “obesity paradox” observed among HF patients [5][6][64].
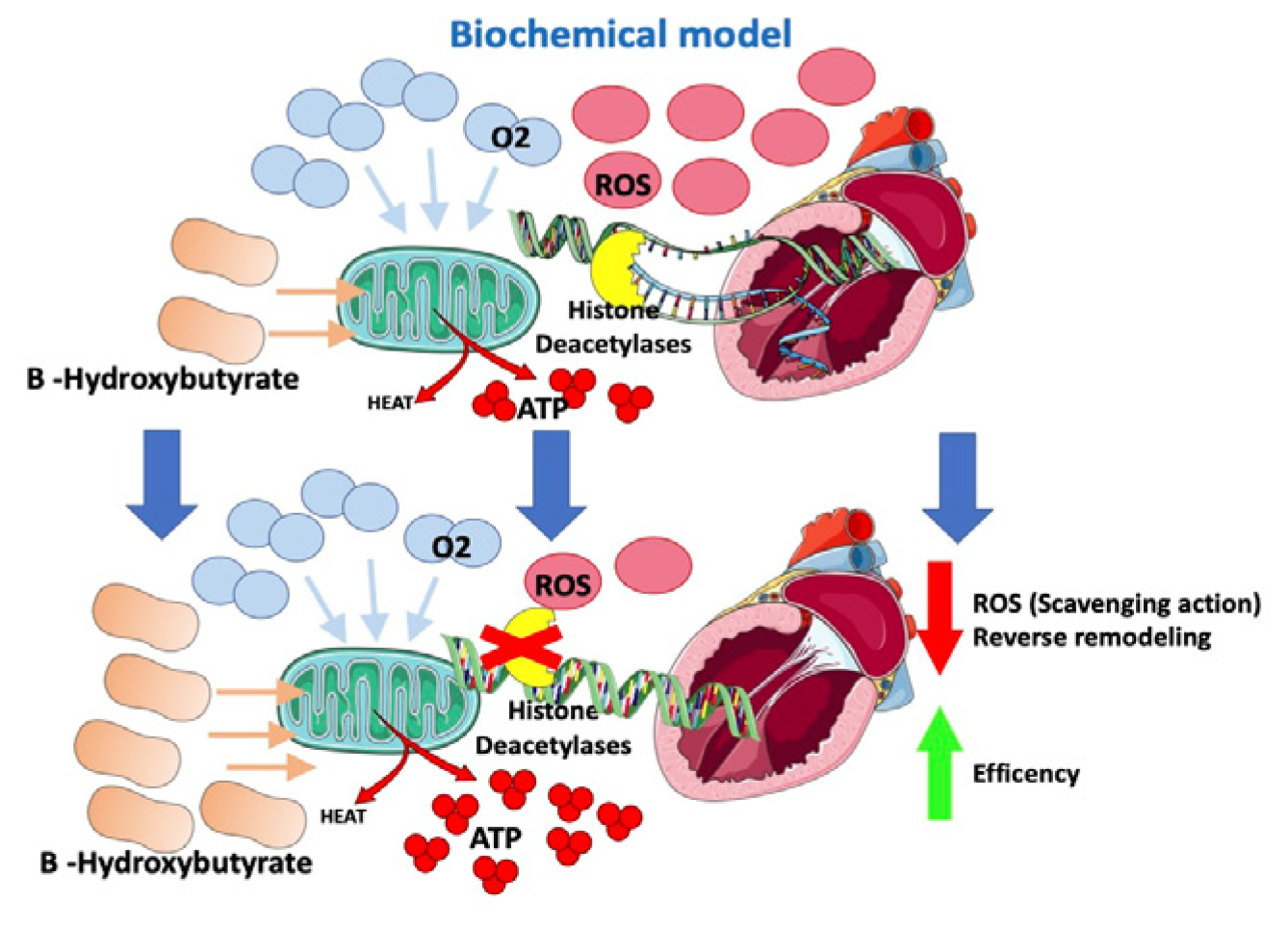
Figure 4. Effects of SGLT2-i on the biochemical model.
The increased ketogenesis improves cardiac function and mechanical efficiency [65]. Empagliflozin improves adverse cardiac remodeling in HF by stimulating the switch of myocardial fuel utilization away from glucose toward ketone bodies, improving myocardial energetics, improving systolic function and getting better cardiac reverse remodeling [66].
The results of the EMPA-VISION trial [67], a double-blind, randomized, placebo-controlled, mechanistic study with the aim of determining the effects of empagliflozin treatment on cardiac energy metabolism and physiology using magnetic resonance spectroscopy (MRS) and cardiovascular magnetic resonance (CMR), have not yet been published. However, due to the suspension of face-to-face contact imposed in March 2020 to restrict transmission of COVID-19, the number of patients included in the analysis of efficacy for the HFpEF was substantially reduced, and this cohort was also underpowered for the planned analysis of the primary endpoint.
In the DEFINE-HF (Dapagliflozin Effects on Biomarkers, Symptoms and Functional Status in Patients With HF With Reduced Ejection Fraction) trial [68], in HFrEF patients, dapagliflozin increased ketone-related and short-chain acylcarnitine as well as medium-chain acylcarnitine principal components analysis-defined metabolite clusters compared with placebo. Changes in metabolites were related to changes in Kansas City Cardiomyopathy Questionnaire scores (i.e., worse quality of life) and in NT-proBNP levels, without interaction by treatment group.
Gliflozins improve heart energetics through an increase in B-hydroxybutyrate, which allows greater ATP production at the cost of lower O2 consumption. Moreover, B-hydroxybutyrate has oxidative stress scavenging action and inhibits histone deacetylases, suppressing the transcription of genes involved in heart hypertrophy and fibrosis.
ROS: reactive oxygen species; ATP: adenosine triphosphate; O2: oxygen.
This entry is adapted from the peer-reviewed paper 10.3390/jcm12010379
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726.
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98.
- Chopra, V.K.; Anker, S.D. Anaemia, iron deficiency and heart failure in 2020: Facts and numbers. ESC Heart Fail. 2020, 7, 2007–2011.
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356.
- Correale, M.; Paolillo, S.; Mercurio, V.; Limongelli, G.; Barillà, F.; Ruocco, G.; Palazzuoli, A.; Scrutinio, D.; Lagioia, R.; Lombardi, C.; et al. Comorbidities in chronic heart failure: An update from Italian Society of Cardiology (SIC) Working Group on Heart Failure. Eur. J. Intern. Med. 2020, 71, 23–31.
- Vest, A.R.; Chan, M.; Deswal, A.; Givertz, M.M.; Lekavich, C.; Lennie, T.; Litwin, S.E.; Parsly, L.; Rodgers, J.E.; Rich, M.W.; et al. Nutrition, Obesity, and Cachexia in Patients With Heart Failure: A Consensus Statement from the Heart Failure Society of America Scientific Statements Committee. J. Card. Fail. 2019, 25, 380–400.
- Triposkiadis, F.; Butler, J.; Abboud, F.M.; Armstrong, P.W.; Adamopoulos, S.; Atherton, J.J.; Backs, J.; Bauersachs, J.; Burkhoff, D.; Bonow, R.O.; et al. The continuous heart failure spectrum: Moving beyond an ejection fraction classification. Eur. Heart J. 2019, 40, 2155–2163.
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378.
- Lüscher, T.F. Heart failure: The cardiovascular epidemic of the 21st century. Eur. Heart J. 2015, 36, 395–397.
- Fonarow, G.C.; Albert, N.M.; Curtis, A.B.; Stough, W.G.; Gheorghiade, M.; Heywood, J.T.; McBride, M.L.; Inge, P.J.; Mehra, M.R.; O’Connor, C.M.; et al. Improving evidence-based care for heart failure in outpatient cardiology practices: Primary results of the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Circulation 2010, 122, 585–596.
- Crespo-Leiro, M.G.; Anker, S.D.; Maggioni, A.P.; Coats, A.J.; Filippatos, G.; Ruschitzka, F.; Ferrari, R.; Piepoli, M.F.; Delgado Jimenez, J.F.; Metra, M.; et al. European Society of Cardiology Heart Failure Long-Term Registry (ESC-HF-LT): 1-year follow-up outcomes and differences across regions. Eur. J. Heart Fail. 2016, 18, 613–625.
- Baldasseroni, S.; Opasich, C.; Gorini, M.; Lucci, D.; Marchionni, N.; Marini, M.; Campana, C.; Perini, G.; Deorsola, A.; Masotti, G.; et al. Left bundle-branch block is associated with increased 1-year sudden and total mortality rate in 5517 outpatients with congestive heart failure: A report from the Italian network on congestive heart failure. Am. Heart J. 2002, 143, 398–405.
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Ben Gal, T.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576.
- Nijst, P.; Verbrugge, F.H.; Grieten, L.; Dupont, M.; Steels, P.; Tang WH, W.; Mullens, W. The pathophysiological role of interstitial sodium in heart failure. J. Am. Coll. Card. 2015, 65, 378–388.
- Swedberg, K. Importance of neuroendocrine activation in chronic heart failure. Impact on treatment strategies. Eur. J. Heart Fail. 2000, 2, 229–233.
- Packer, M. The neurohormonal hypothesis: A theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Card. 1992, 20, 248–254.
- Mann, D.L. Progress in Pediatric Cardiology The evolution of modern theory and therapy for heart failure. Prog. Pediatr. Cardiol. 2014, 37, 9–12.
- Fish-Trotter, H.; Ferguson, J.F.; Patel, N.; Arora, P.; Allen, N.B.; Bachmann, K.N.; Daniels, L.B.; Reilly, M.P.; Lima, J.A.C.; Wang, T.J.; et al. Inflammation and Circulating Natriuretic Peptide Levels. Circ. Heart Fail. 2020, 13, e006570.
- von Haehling, S.; Ebner, N.; Dos Santos, M.R.; Springer, J.; Anker, S.D. Muscle wasting and cachexia in heart failure: Mechanisms and therapies. Nat. Rev. Cardiol. 2017, 14, 323–341.
- Thandavarayan, R.A.; Chitturi, K.R.; Guha, A. Pathophysiology of Acute and Chronic Right Heart Failure. Cardiol. Clin. 2020, 38, 149–160.
- Tóth, M.; Vuorinen, K.H.; Vuolteenaho, O.; Hassinen, I.E.; Uusimaa, P.A.; Leppäluoto, J.; Ruskoaho, H. Hypoxia stimulates release of ANP and BNP from perfused rat ventricular myocardium. Am. J. Physiol. 1994, 266 Pt 2, H1572–H1580.
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242.
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271.
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90.
- Katsuno, K.; Fujimori, Y.; Takemura, Y.; Hiratochi, M.; Itoh, F.; Komatsu, Y.; Fujikura, H.; Isaji, M. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J. Pharmacol. Exp. Ther. 2007, 320, 323–330.
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117.
- Wanner, C.; Lachin, J.M.; Inzucchi, S.E.; Fitchett, D.; Mattheus, M.; George, J.; Woerle, H.J.; Broedl, U.C.; von Eynatten, M.; Zinman, B.; et al. Empagliflozin and Clinical Outcomes in Patients With Type 2 Diabetes Mellitus, Established Cardiovascular Disease, and Chronic Kidney Disease. Circulation 2018, 137, 119–129.
- Verma, S.; McMurray, J.J.V.; Cherney, D.Z.I. The Metabolodiuretic Promise of Sodium-Dependent Glucose Cotransporter 2 Inhibition: The Search for the Sweet Spot in Heart Failure. JAMA Cardiol. 2017, 2, 939–940.
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018, 41, 356–363.
- Kimura, T.; Sanada, J.; Shimoda, M.; Hirukawa, H.; Fushimi, Y.; Nishioka, M.; Kinoshita, T.; Okauchi, S.; Obata, A.; Kohara, K.; et al. Switching from low-dose thiazide diuretics to sodium-glucose cotransporter 2 inhibitor improves various metabolic parameters without affecting blood pressure in patients with type 2 diabetes and hypertension. J. Diabetes Investig. 2018, 9, 875–881.
- Lytvyn, Y.; Bjornstad, P.; Udell, J.A.; Lovshin, J.A.; Cherney, D.Z.I. Sodium Glucose Cotransporter-2 Inhibition in Heart Failure: Potential Mechanisms, Clinical Applications, and Summary of Clinical Trials. Circulation 2017, 136, 1643–1658.
- Matthews, V.B.; Elliot, R.H.; Rudnicka, C.; Hricova, J.; Herat, L.; Schlaich, M.P. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J. Hypertens. 2017, 35, 2059–2068.
- Nguyen, T.; Wen, S.; Gong, M.; Yuan, X.; Xu, D.; Wang, C.; Jin, J.; Zhou, L. Dapagliflozin Activates Neurons in the Central Nervous System and Regulates Cardiovascular Activity by Inhibiting SGLT-2 in Mice. Diabetes Metab. Syndr. Obes. 2020, 13, 2781–2799.
- McMurray, J. EMPA-REG—The “diuretic hypothesis”. J. Diabetes Complicat. 2016, 30, 3–4.
- Scheen, A.J. Effect of SGLT2 Inhibitors on the Sympathetic Nervous System and Blood Pressure. Curr. Cardiol. Rep. 2019, 21, 70.
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487.
- Fedak, P.W.M.; Verma, S.; Weisel, R.D.; Li, R.-K. Cardiac remodeling and failure: From molecules to man (Part I). Cardiovasc. Pathol. 2005, 14, 1–11.
- Correale, M.; Mazzeo, P.; Tricarico, L.; Croella, F.; Fortunato, M.; Magnesa, M.; Amatruda, M.; Alfieri, S.; Ferrara, S.; Ceci, V.; et al. Pharmacological Anti-Remodelling Effects of Disease-Modifying Drugs in Heart Failure with Reduced Ejection Fraction. Clin. Drug Investig. 2022, 42, 567–579.
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients With Heart Failure: Proposal of a Novel Mechanism of Action. JAMA Cardiol. 2017, 2, 1025–1029.
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726.
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573.
- Garvey, W.T.; Van Gaal, L.; Leiter, L.A.; Vijapurkar, U.; List, J.; Cuddihy, R.; Ren, J.; Davies, M.J. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 2018, 85, 32–37.
- Fedak, P.W.; Verma, S.; Weisel, R.D.; Li, R.K. Cardiac remodeling and failure: From molecules to man (Part II). Cardiovasc. Pathol. 2005, 14, 49–60.
- Fedak, P.W.; Verma, S.; Weisel, R.D.; Skrtic, M.; Li, R.K. Cardiac remodeling and failure: From molecules to man (Part III). Cardiovasc. Pathol. 2005, 14, 109–119.
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310.
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.J.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553.
- Packer, M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis. Diabetes Obes. Metab. 2018, 20, 1361–1366.
- Requena-Ibáñez, J.A.; Santos-Gallego, C.G.; Rodriguez-Cordero, A.; Vargas-Delgado, A.P.; Mancini, D.; Sartori, S.; Atallah-Lajam, F.; Giannarelli, C.; Macaluso, F.; Lala, A.; et al. Mechanistic Insights of Empagliflozin in Nondiabetic Patients With HFrEF: From the EMPA-TROPISM Study. JACC Heart Fail. 2021, 9, 578–589.
- Neeland, I.J.; Yokoo, T.; Leinhard, O.D.; Lavie, C.J. 21st Century Advances in Multimodality Imaging of Obesity for Care of the Cardiovascular Patient. JACC Cardiovasc. Imaging 2021, 14, 482–494.
- Braunwald, E. Gliflozins in the Management of Cardiovascular Disease. N. Engl. J. Med. 2022, 386, 2024–2034.
- Elrakaybi, A.; Laubner, K.; Zhou, Q.; Hug, M.J.; Seufert, J. Cardiovascular protection by SGLT2 inhibitors—Do anti-inflammatory mechanisms play a role? Mol. Metab. 2022, 64, 101549.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128.
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.M.; Hantel, S.; Salsali, A.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Inzucchi, S.E.; et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: Results of the EMPA-REG OUTCOME® trial. Eur. Heart J. 2016, 37, 1526–1534.
- Tikkanen, I.; Narko, K.; Zeller, C.; Green, A.; Salsali, A.; Broedl, U.C.; Woerle, H.J. Empagliflozin Reduces Blood Pressure in Patients With Type 2 Diabetes and Hypertension. Diabetes Care 2015, 38, 420–428.
- Li, H.; Shin, S.E.; Seo, M.S.; An, J.R.; Choi, I.W.; Jung, W.K.; Firth, A.L.; Lee, D.S.; Yim, M.J.; Choi, G.; et al. The anti-diabetic drug dapagliflozin induces vasodilation via activation of PKG and Kv channels. Life Sci. 2018, 197, 46–55.
- Kolwelter, J.; Bosch, A.; Jung, S.; Stabel, L.; Kannenkeril, D.; Ott, C.; Bramlage, P.; Schiffer, M.; Achenbach, S.; Schmieder, R.E. Effects of the sodium-glucose cotransporter 2 inhibitor empagliflozin on vascular function in patients with chronic heart failure. ESC Heart Fail. 2021, 8, 5327–5337.
- Correale, M.; Mazzeo, P.; Mallardi, A.; Leopizzi, A.; Tricarico, L.; Fortunato, M.; Magnesa, M.; Tucci, S.; Maiellaro, P.; Pastore, G.; et al. Switch to SGLT2 Inhibitors and Improved Endothelial Function in Diabetic Patients with Chronic Heart Failure. Cardiovasc. Drugs Ther. 2022, 36, 1157–1164.
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644.
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716.
- Kolwicz, S.C.J.; Airhart, S.; Tian, R. Ketones Step to the Plate: A Game Changer for Metabolic Remodeling in Heart Failure? Circulation 2016, 133, 689–691.
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114.
- Lopaschuk, G.D.; Verma, S. Empagliflozin’s Fuel Hypothesis: Not so Soon. Cell Metab. 2016, 24, 200–202.
- Santos-Gallego, C.G.; Ibanez, J.A.R.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste Botija, M.B.; Sanz Salvo, A.J.; Hajjar, R.; Fuster, V.; Badimon, J. Empagliflozin induces a myocardial metabolic shift from glucose consumption to ketone metabolism that mitigates adverse cardiac remodeling and improves myocardial contractility. J. Am. Coll. Cardiol. 2018, 71, A674.
- Carbone, S.; Lavie, C.J.; Arena, R. Obesity and Heart Failure: Focus on the Obesity Paradox. Mayo Clin. Proc. 2017, 92, 266–279.
- Mizuno, Y.; Harada, E.; Nakagawa, H.; Morikawa, Y.; Shono, M.; Kugimiya, F.; Yoshimura, M.; Yasue, H. The diabetic heart utilizes ketone bodies as an energy source. Metabolism 2017, 77, 65–72.
- Correale, M.; Lamacchia, O.; Ciccarelli, M.; Dattilo, G.; Tricarico, L.; Brunetti, N.D. Vascular and metabolic effects of SGLT2i and GLP-1 in heart failure patients. Heart Fail. Rev. 2021. online ahead of print.
- Hundertmark, M.J.; Agbaje, O.F.; Coleman, R.; George, J.T.; Grempler, R.; Holman, R.R.; Lamlum, H.; Lee, J.; Milton, J.E.; Niessen, H.G.; et al. Design and rationale of the EMPA-VISION trial: Investigating the metabolic effects of empagliflozin in patients with heart failure. ESC Heart Fail. 2021, 8, 2580–2590.
- Selvaraj, S.; Fu, Z.; Jones, P.; Kwee, L.C.; Windsor, S.L.; Ilkayeva, O.; Newgard, C.B.; Margulies, K.B.; Husain, M.; Inzucchi, S.E.; et al. Metabolomic Profiling of the Effects of Dapagliflozin in Heart Failure With Reduced Ejection Fraction: DEFINE-HF. Circulation 2022, 146, 808–818.
This entry is offline, you can click here to edit this entry!