Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Ecology
The phenotypic diversity required for environmental adaptation can be produced by genetic and epigenetic mechanisms.
- asexual populations
- epigenetic ecotypes
- ecoepigenetics
1. Generation of Phenotypic Variation by Genetic Mechanisms
Genetic mutations such as single nucleotide substitutions, duplications, deletions and transversions are fundamental processes for the generation of phenotypic diversity. Random single nucleotide substitutions have a frequency of 10−8–10−9 per locus and generation for most organisms [26]. The genetic diversity that can be generated by this mechanism is very much dependent on population size and generation time. Populations with low individual numbers and long generation times can produce much less genetic variation than populations with high individual numbers and short generation times. Since most mutations are deleterious or neutral only a smaller proportion contributes to phenotypic variation in a population. Previously, genetic mutations were thought to occur randomly throughout the genome, but this has been challenged in recent years. For example, in the model plant Arabidopsis thaliana, mutations were less frequently found in functionally constrained regions of the genome [27]. In gene bodies the mutation rate was reduced by half and in essential genes by two-thirds.
Recombination, the exchange of DNA between maternal and paternal chromosomes during meiosis that produces new allele combinations and phenotypic variants, is typical of sexually reproducing animals and plants. It is variable between higher taxa and species, variable across the genome, and highest under conditions of random mating [28]. The effects of recombination can be positive, facilitating environmental adaptation, or negative, breaking apart beneficial allele combinations. Only sexually reproducing organisms can use recombination to produce phenotypic variation in populations. There are also possibilities of horizontal gene transfer in bacteria, which are summarized under the term recombination [29].
Genetic drift, the random change in the frequency of an allele from generation to generation, may influence the spectrum of phenotypic diversity in a population by either causing gene variants to disappear or causing initially rare alleles to become frequent. The role of drift is expected to be particularly important in small and isolated populations [30]. Gene flow, the transfer of genetic material from one population to another, can also considerably influence phenotypic diversity in a population [31].
2. Generation of Phenotypic Variation by Epigenetic Mechanisms
The generation of phenotypic diversity from the same genome by epigenetic mechanisms is most easily investigated by performing experiments with genetically identical clone members [32,33]. Suitable animal models are monozygotic twins and polyembryonic multiples of mammals, clone-mates of apomictic parthenogenetic vertebrates and invertebrates, and genets of colonial corals. Good plant models are individuals of clonal lineages and cuttings of the same individual, and good microbial models are yeast and bacterial colonies originating from a single founder cell by binary fission.
The best investigated epigenetic mechanisms that are known to be involved in the generation of phenotypic diversity from the same genome are DNA methylation, histone modifications and non-coding RNAs [34,35,36]. Recent reviews of epigenetic mechanisms in animals, plants, fungi and bacteria are provided by Vogt [37], Maeji and Nishimura [38], Madhani [39] and Sánchez-Romero and Casadesús [40], respectively. The full range of epigenetic chromatin modifications in an organism that can change gene expression and contribute to phenotypic variation is currently unknown. However, the diploid human epigenome contains >107 CpG dinucleotides and >108 histone tails, providing an enormous number of potentially modifiable sites [41]. The number of human microRNA genes, each of which can produce numerous transcripts involved in the generation of phenotypic variation is approximately 2600 [42].
DNA methylation is the best investigated epigenetic mechanism. It is widespread in prokaryotes and eukaryotes but has been lost in some species and groups [37,43]. In animals, the methylation marks are mostly on the cytosines of CpG dinucleotides and occur in genic and intergenic regions including promoters, gene bodies and repeats [34,44,45]. Methylation of promoters and transposons usually results in transcriptional repression, whereas gene body methylation modifies the accessibility of genes in the chromatin, modulates gene expression, and prevents spurious transcription initiation [45,46,47]. The DNA methylation marks in animals are established by DNA methyltransferases (DNMTs) and erased by ten-eleven-translocation enzymes (TETs) [48,49].
In plants, DNA methylation is found in CG, CHG and CHH sequence contexts, where H is A, C or T [50]. Methylation is highly enriched in repressed transposable elements and repeats. Plants have different methylation and demethylation enzymes when compared to animals [50]. In fungi, DNA methylation is mainly found in transposable elements, promoter regions and repetitive DNA sequences [51,52]. It is associated with silencing of gene expression and transposons and is involved in a wide range of biological phenomena including phenotypic switching. Bacterial genomes mainly display adenine methylation, which helps regulating numerous cellular processes such as chromosome replication, correction of DNA mismatches and transcription [53]. It is further involved in bacterial defence and virulence and fosters formation of phenotypically different epigenetic lineages.
Post-translational histone modifications are typical of all eukaryotes [35,54]. The histones in the nucleosomes greatly influence DNA transcription by either shielding the DNA or allowing binding of transcription factors to the DNA. In animals, the N-terminal tails of the histones carry modifications such as methylation, acetylation, phosphorylation and ubiquitination, which affect the chromatin structure. Histone acetylation often stimulates gene expression, whereas histone methylation often represses gene expression, depending on the amino acid residue being modified. The histone modifications are produced by a broad array of enzymes and read by various proteins [55,56]. For example, histone acetylation marks in animals are written by histone acetyltransferases (HATs) and read by bromodomain-containing proteins (BrDs). Information on histone modifications and their regulation in plants and fungi is found in Zhao et al. [57] and Brosch et al. [58], respectively.
Non-coding RNAs are further crucial regulators of gene expression and contributors to phenotypic variation that occur in eukaryotes and prokaryotes [36,59]. They include several classes that differ in sequence length and molecular configuration. In animals, microRNAs inhibit translation or cause mRNA degradation [60,61]. Small interfering RNAs regulate gene transcription through transposable element silencing and the interaction with DNA methylation and histone modifications [62]. Piwi-interacting RNAs mainly silence transposable elements in the germ line at the transcriptional and post-transcriptional levels [63]. Long ncRNAs are involved in transcriptional regulation, dosage compensation and genomic imprinting in mammals and development, insecticide resistance and anti-viral defence in insects [64,65]. Information on the role of non-coding RNAs in plants, fungi and bacteria is found in Waititu et al. [66], Dhingra [67] and Stav et al. [68], respectively.
Polycomb group (PcG) and Trithorax group (TrxG) proteins contribute significantly to the mitotic and meiotic inheritance of epigenetically mediated phenotypic variability in eukaryotes by sustaining silent and active gene expression states through cell generations and the germ line [69]. Ciabrelli et al. [70] demonstrated for isogenic lines of the fruit fly Drosophila melanogaster how chromatin organization and PcG proteins support epigenetically heritable phenotypic plasticity.
Phenotypic variation unrelated to genetic variation can additionally be produced by alternative splicing, RNA editing and chemical modifications of the mRNA. Alternative splicing generates multiple transcripts from a single gene. It is basically a genetic mechanism [71], but epigenetic mechanisms can be involved as well. For example, methylation of CpGs and histone modifications can mark an alternative exon, and these marks are then recognized by an adaptor protein that recruits the splicing factors [72]. An example of mRNA editing is the deamination of adenosine to inosine by the ADAR (adenosine deaminase acting on RNA) enzyme family, which can lead to codon change [73]. Chemical modifications of the mRNA can result in codon change and diversification of the proteome and phenome as well [74].
3. Stochastic and Environmentally-Induced Epimutations and Related Phenotypic Change
Changes of the epigenetic marks on the DNA and histones that can trigger phenotypic variation occur spontaneously or by environmental induction [32,33,75,76,77]. These changes are called epimutations [78]. Epimutations do not alter the DNA sequence and are principally reversible. Like genetic mutations, stochastic epimutations affect first only single individuals of a population. In genetically identical populations, all stochastic epimutations together can generate a phenotypic spectrum around the target or mean phenotype [79,80]. When the environment changes, one of these alternative phenotypes may become the optimal one. Thus, the a-priori production of a spectrum of phenotypic variants from the same genotype by stochastic epimutations without knowing the future conditions can be regarded as a bet-hedging strategy that secures populations against unforeseen changes of the environment. In contrast, environmentally induced epimutations can affect many population members simultaneously, strengthening adaptation to the prevailing conditions and creating phenotypic stability. In wild populations, both sources of epigenetic variation occur together [80] as will be discussed in more detail below.
Stochastic epimutations can be neutral, detrimental or beneficial such as genetic mutations. However, they are several orders of magnitude more frequent than genetic mutations. For example, in the model plant Arabidopsis thaliana, stochastic epimutations occur at a rate of 10−4 per base pair and generation, whilst genetic mutations occur only at a rate of 10−9 [81].
The environmental induction of phenotypic change via epigenetically-mediated differential gene expression is triggered by stronger and longer lasting environmental cues such as temperature, drought, light, salinity, food, predator odours, injury, toxicants or disease agents [82,83,84]. It requires signal transmission from the external world to the nucleus of the target cells, environment-sensitive molecules involved in gene regulation that can perceive and interpret these signals, readers and editors of epigenetic marks, and molecules that recruit the epigenetic modifiers to specific regions of the DNA and chromatin. These various components must crosstalk to specifically change expression of a gene (Figure 1).
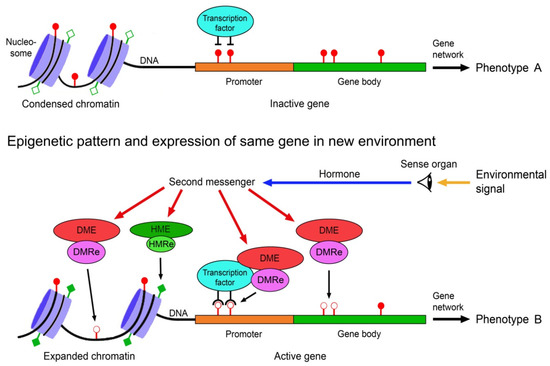
Figure 1. Scheme of environmentally induced change of gene and phenotype expression by epigenetic mechanisms. Environmental signals trigger gene expression change via hormones, second messengers, and environment-sensitive DNA methylation modifying enzymes (DME) and histone modifying enzymes (HME). DNA methylation readers (DMRe), histone modification readers (HMRe) and transcription factors recruit the DMEs and HMEs to specific sites in the chromatin and DNA. Histone modifications such as acetylation (filled squares) and deacetylation (open squares) help to shape chromatin structure and access to the DNA, and methylation (filled circles) and demethylation (open circles) of CpG dinucleotides in the DNA modify gene expression, resulting in different variants of a phenotypic trait. Adapted from Vogt [9].
The transmission of environmental signals to the target cells mostly occurs via signal perceiving sense organs and signal transmitting neurohormones and second messengers, which eventually regulate the molecules involved in chromatin remodelling, gene expression and processing of the transcripts. Serotonin is a good example of an environmental signal transmitting hormone. In locust polyphenism, the density-dependent change of morphologically and behaviourally different stationary and migratory phases, it regulates the alternative expression of density-responsive genes with the help of epigenetic mechanisms [85].
A considerable number of proteins participating in the regulation of chromatin architecture and gene expression are responsive to environmental cues. Examples are the DNA demethylating TET, which is up- or downregulated by several environmental factors including food ingredients, ethanol, air pollution and radiation [86], proteins of the Polycomb group that are sensitive to the environmental temperature [87], and transcription factors of the TCP family in vascular plants that mediate environmental signals into growth responses [88].
The writers and erasers of the DNA methylation marks include the methylating DNMTs and the demethylating TETs [48,49]. These enzymes form complexes with readers of the DNA methylation marks such as proteins of the methyl-CpG-binding domain family (MBDs) and transcription factors to exert their functions [89,90,91]. The MBDs bind methylated CpG dinucleotides and act as translators between DNA methylation and histone modifications [91]. Transcription factors with different sequence specificity can guide the methylation modifying molecular complex to specific sites of the DNA [92]. Each animal and plant possess hundreds of such transcription factors.
This entry is adapted from the peer-reviewed paper 10.3390/epigenomes7010001
This entry is offline, you can click here to edit this entry!