Renal cell carcinoma (RCC) makes up the majority of kidney cancers, with a poor prognosis for metastatic RCC (mRCC). Challenges faced in the management of mRCC, include a lack of reliable prognostic markers and biomarkers for precise monitoring of disease treatment, together with the potential risk of toxicity associated with more recent therapeutic options. Glycosaminoglycans (GAGs) are a class of carbohydrates that can be categorized into four main subclasses, viz., chondroitin sulfate, hyaluronic acid, heparan sulfate and keratan sulfate. GAGs are known to be closely associated with cancer progression and modulation of metastasis by modification of the tumor microenvironment. Alterations of expression, composition and spatiotemporal distribution of GAGs in the extracellular matrix (ECM), dysregulate ECM functions and drive cancer invasion.
1. Metastatic Renal Cell Carcinoma
Renal cell carcinoma (RCC) comprises more than 90% of cases of kidney cancer, with clear cell RCC (ccRCC) being the most common type of RCC and making up the majority of cancer-related deaths [
1,
2]. The “founding event” of ccRCC is often attributed to a mutation in the von Hippel-Lindau (VHL) tumor suppressor gene [
3], although by itself is insufficient to cause ccRCC. The prognosis for RCC is poor, especially for metastatic RCC (mRCC). The overall 5-year survival rate for RCC patients is 74% and decreases to only 8% for patients with mRCC [
4,
5]. Despite improvements in early detection techniques and considerable progress in systemic treatment, a quarter of patients with localized RCC still develops metastatic deposits at distant sites following surgical removal of the primary tumor (post-nephrectomy) [
6,
7]. Distant metastases are mostly observed in the lymph nodes, lungs, liver, bone and brain [
8].
For early or resectable RCC, nephrectomy is usually performed in the management of this cancer. As RCC is usually resistant to conventional chemotherapy and radiotherapy, the standard treatments for mRCC have been interleukin-2 and interferon cytokine-based therapies, until the availability of targeted therapies [
2,
9]. Several targeted treatments are available for mRCC management, the most common being tyrosine kinase inhibitors targeting Vascular endothelial growth factor (VEGF) signaling, such as sunitinib [
10,
11] and sorafenib [
12]. Sunitinib is commonly used as a first line treatment option for RCC, with a higher response rate and longer progression-free survival than the conventionally used interferon α as observed during a Phase 3 clinical trial [
10]. Moreover, poor-risk RCC patients were also observed to show responses to the drug in the same study. Although sunitinib is still used in VEGFR-targeted therapy for advanced RCC [
13], newer generation of multiple VEGF kinase inhibitors, such as Lenvatinib, has been found to be more effective [
14].
Another drug target is the mammalian target of rapamycin (mTOR) pathway, which regulates cell proliferation and tumor metabolism [
15]. Temsirolimus, a specific inhibitor of mTOR, has been used as both first line and second line treatment options in advanced RCC [
16,
17]. This intravenous drug has also been shown to achieve prolonged survival over interferon α among mRCC patients in a Phase 3 clinical trial [
18,
19]. However, despite the clinically beneficial outcomes that these targeted treatments offer, nearly all RCC patients develop resistance to both VEGF-targeted and mTOR-targeted therapies. Thus, a combination of VEGF and mTOR inhibitors has been administered as a strategy to delay drug resistance to either class of the inhibitors [
20,
21].
Another treatment modality employed is immunotherapy targeting the programmed cell death protein 1 (PD1) and its ligand PDL1, as demonstrated by the effects of the drug nivolumab [
22]. PDL1 is overexpressed in cancer cells, and inhibiting PD1-PDL1 interaction promotes T-cell activation and killing of cancer cells. Nivolumab, used as second line treatment for mRCC, has been reported to offer a longer overall survival and higher response rates, with fewer adverse effects and a better quality of life, compared to the mTOR inhibitor Everolimus [
23]. A meta-analysis comprising 5121 patients with mRCC from six clinical trials, revealed that Nivolumab plus cabozantinib (an oral inhibitor of multiple tyrosine kinases) was associated with the highest likelihood of patients having maximal overall survival, while the combination of Lenvatinib plus Pembrolizumab (a humanized antibody used in cancer immunotherapy), the highest likelihood of progression free survival [
24]. In fact, the European Association of Urology Guidelines has recommended combination therapies of Axitinib plus Pembrolizumab, Cabozantinib plus Nivolumab, and Lenvatinib plus Pembrolizumab for advanced RCC [
14]. Another combination therapy which has undergone a phase III clinical trial include, randomization of 873 patients who received either axitinib and avelumab or sunitinib [
25]. A recent report has also shown evidence for clinically meaningful and durable benefits in advanced RCC patients treated with Nivolumab plus Ipilimumab (a monoclonal antibody that targets CTLA-4) [
26].
2. Glycosaminoglycans
Glycosaminoglycans (GAGS) are linear polysaccharides that consist of repeating disaccharide units of uronic acid and an amino sugar [
33]. They are found in almost every mammalian tissue, providing structural scaffolding and hydration to the cells [
34].
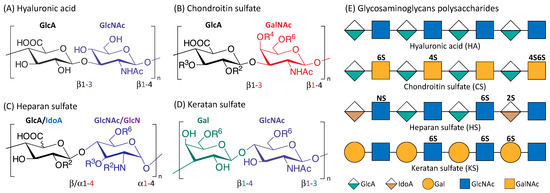
Figure 1 is an illustration of the four main classes of glycosaminoglycans: hyaluronic acid (HA), chondroitin sulfate (CS), heparan sulfate (HS), and keratan sulfate (KS). Monomers of the disaccharide building blocks consist of GlcA (d-glucuronic acid) and GlcNAc (
N-acetyl-d-glucosamine) for HA; GlcA and GalNAc (
N-acetyl-d-galactosamine) for CS; GlcA or IdoA (l-iduronic acid) and GlcNAc or GlcN (d-glucosamine) for HS; Gal (d-galactose) and GlcNAc for KS. GAGs are highly polar and negatively charged with the polysaccharide lengths generally varying between 4 and 200 mer [
35]. With the exception of HA, GAGs contain sulfate groups attached at specific sites [
36]. The sulfate groups are added onto the GAGs chain through post-polymerization modifications [
37]. O-sulfotransferases mediate the sulfation of CS and KS while the sulfation of HS is controlled by N-sulfotransferases, C5 epimerases as well as O-sulfotransferases [
35,
38].
Figure 1. Structure of the four main glycosaminoglycans (GAGs). Disaccharide monomers of Hyaluronic acid (A); Chondroitin sulfate (B); Heparan sulfate (C) and Keratan sulfate (D). The possible sulfate sites are denoted with Ri, the superscript ‘i’ indicate the Carbon position where the sulfate group is esterified; R = H or SO3H. Representative Glycosaminoglycans polysaccharides of HA, CS, HS and KS consist of repeating disaccharides monomers with various sulfation patterns (E).
HA (the only GAG known not to have any sulfation sites) has a crucial role in cushioning and lubricating the body that is attributable to its highly hydrophilic property, and is therefore found in abundance in the eyes, joints, and heart valves [
39]. HA is also abundant in the skin and important in wound healing [
40]. KS is present in the cornea, cartilage, and bones, and associated with disorders such as macular corneal dystrophy and osteoarthritis [
41]. HS is usually located in the extracellular matrix (ECM), and is highly involved in tumorigenesis [
42,
43,
44]. CS is an important structural component of cartilage, providing much of their resistance to compression [
45]. CS can interact with various biomolecules and form proteoglycans (PGs) with proteins, the major components of the extracellular matrix and drive crucial biological activities. The various sulfation patterns of CS could code for different biological regulatory functions [
46,
47]. For instance, C4S is known to be important for cartilage regeneration and observed to be downregulated in degraded osteoarthritic cartilage [
48]. C4,6S augmented cartilage generation through enhancing type II collagen production [
49]. C4,6S, but not C4S or C6S, was reported to be able to interact with several neurotrophic factors to stimulate neurite outgrowth [
46].
Interestingly, it is well established that GAGs are involved in cancer cell growth, signalling, and metastasis [
34,
50]. HA levels are significantly elevated in breast [
51], lung [
52], and ovarian cancers [
53]. Certain sulfation motifs of exogenous CS were shown to induce apoptosis and inhibit the growth of triple negative breast cancer cells [
54]. Dermatan sulfate was observed to be elevated with changes in the sulfation profiles in the stroma of certain cancers such as liver [
55], lung [
56,
57], pancreatic [
58], colorectal [
59] and gastric [
60] cancers.
3. GAGs and Metastasis
Cancer is a complex disease where malignant cells could acquire the ability to metastasize to distant sites, thus accounting for the majority of cancer-related morbidity and mortality. The fundamental processes of migration and invasion are crucial for cancer metastasis, which is usually the primary cause of death in cancer patients [
61]. The ECM, which is composed mainly of GAGs and their PGs, would therefore play a significant role in controlling cell behavior and movement [
62]. GAGs are endowed with rigidity property, thus providing structural integrity to the cells and passageways in the ECM between cells [
36]. Dysregulation of ECM modelling occurs during tumorigenesis and metastasis, leading to changes in the tumor microenvironment (TME) and loss of tissue homeostasis [
63,
64]. Disorganization in the ECM GAGs/PGs expression, composition and spatiotemporal distribution are the main causes of the dysregulation of ECM functions and the driver for cancer invasion [
65]. GAGs modulate cancer invasion through binding with various growth factors, adhesion molecules and cytokines [
66].
HA is the only GAG that does not form PGs with any protein through covalent bonding, and therefore not sulfated at all [
37]. Increased HA synthesis through overexpression of Hyaluronan Synthases (HS) was observed to promote cancer growth and metastasis in xenograft models of breast, prostate, and colon cancers [
67,
68,
69,
70]. The presence of HA provides cancer cells with a highly hydrated and malleable ECM which is essential for changes in cell shapes and tissue penetration during invasion [
71,
72,
73]. Pericellular HA surrounding metastatic cancer cells could facilitate adhesion of cancer cells to endothelial cells at the metastatic site [
74,
75] (
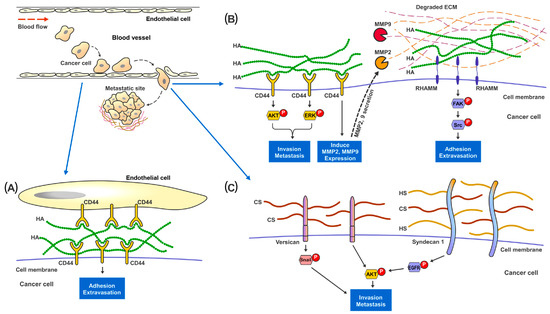
Figure 2). Moreover, HA can interact with various cell-surface receptors, notably CD44 and RHAMM, which are well established to be involved in cancer cell survival, motility, and metastasis [
76,
77]. Studies have shown that disruption of the binding of HA to either CD44 or RHAMM receptors [
78,
79,
80], would suppress the development of metastatic nodules in mice. HA-CD44 interaction has been reported to stimulate Matrix Metalloproteinase 2 (MMP2) and MMP9 expression and their cell-surface presentation [
81]. These MMPs play important roles in cancer invasion, as they aid in digesting through the ECM barrier (which is essential in preventing cancer cells from escaping their primary tissue architecture) and in facilitating the growth of cancer cells at metastatic sites [
82,
83]. HA bound to RHAMM could induce the activation of FAK which is required for actin filament and microtubule rearrangements as well as cancer cell motility [
84,
85,
86,
87].
Figure 2. Proposed mechanisms of the involvement of GAGs in cancer metastasis. In metastatic renal cancer, HA, HS and HS present on the cell surface layer promote cancer cell invasion and metastatic capabilities. (A) During metastasis, cancer cells traveling in the blood vessel adhere to the endothelial cells through the binding and interaction between cell surface CD44 and HA present in the ECM. (B) HA-bound CD44 receptors on cancer cells activates MAPK-ERK1/2 and AKT signaling pathways to promote cell adhesion, migration and invasion. CD44 also increases MMP2 and MMP9 expression and secretion to help digest and remodel ECM proteins at the metastatic site. Cell surface RHAMM, upon the binding of HA in the ECM, activates FAK-Src signaling pathways to help cancer cells migrate through the blood vessel and start colonization at the metastatic site. (C) Cell surface proteoglycan of CS and HS, such as Versican and Syndecan-1, promote cancer invasion though activating Snail, EGFR and AKT signaling pathways.
CS, HS and KS can be sulfated and form PGs with the core proteins through covalent binding at the Serine residues [
88]. Alterations in cell surface CS expression, sulfation patterns and consequently, ECM-degradative enzymes, such as MMPs, would result in changes of cell invasiveness and disruption of cell-matrix interactions [
33,
89]. In vitro studies on breast cancer revealed that a higher CS expression in tumor cells was concomitant with increased cell proliferation, migration and invasion [
90,
91,
92,
93]. At the tissue level, CS in general was observed to be significantly elevated in the stromal compartment of breast tumors [
94,
95]. The sulfation patterns of CS also elicited various effects on cancer invasion [
89]. For instance, elevated expression of non-sulfated chondroitin in prostate cancer was associated with adverse clinicopathological parameters [
96]. C4,6S was noted to suppress cancer cell invasion through inhibiting Wnt/β- Catenin signaling [
97] and enhancing the retention of tissue inhibitor of metalloproteinases (TIMP)-3, which disrupt ECM processing and cell mobility [
98]. On the other hand, C4,6S expression could promote ovarian cancer metastasis through interacting with VEGF, HGF [
99,
100] and P-Selectin [
101], which support survival of the circulating cancer cells and tissue colonization. C6S bound to CD44 has been reported to promote cancer cell adhesion and migration [
102]. C4S is known to suppress cancer invasiveness and inhibit cathepsin S activity which regulate cell–cell and cell-ECM contacts [
103,
104]. CS proteoglycans (CSPGs) also participate in cancer cells migration, invasion, and metastasis. A large ECM CSPG (Versican) was observed to promote cancer epithelial-to-mesenchymal transition (EMT) and metastasis through EGFR/AKT [
105], Snail/PAPSS2 [
106] and TGFβ/NK-κB signaling [
107] in liver, breast and ovarian cancers. Intracellular CSPG Serglycin has been reported to interact with CD44 [
108,
109] and activate IL-8 [
110] signaling pathways to enhance cell migration and metastasis in lung and breast cancer.
HS is present at the ECM interface to modulate various types of cell-ECM interactions [
111]. The capability of HS to bind to various chemokines, growth factors, morphogens, enzymes and ECM proteins, confer functional properties such as controlling cancer migration, EMT and metastasis [
34]. Changes in HS sulfation patterns could also affect cancer cell invasion and metastasis [
112,
113]. Reduction in 6-O-Sulfation of HS has been observed to augment VEGF and FGF induced cell invasion in RCC [
114]. On the other hand, an increase in 3-O-Sulfation could enhance the EMT and invasion capacity of pancreatic cancer cells [
115]. Cell surface HS/CS Proteoglycans (HSPGs) Syndecan-1 is known to increase cancer stemness and invasiveness through stimulating the Notch and EGFR signaling pathways and regulation of the focal adhesion kinase-Wnt signaling axis [
116,
117]. In contrast, ECM HSPG Perlecan has been shown to inhibit cancer cell invasion and digested by MMP7 during FAK driven invasion in prostate cancer [
118].
KS expression was reported to be increased in pancreatic tumor tissues compared to normal adjacent tissues and stroma, with KS expression being higher in lung metastatic sites compared to the primary pancreatic tumor [
119]. KSPG Lumican has been shown to inhibit lung cancer invasion through binding with p120-catenin, which prevent activation of Rho GTPases, FAK and cytoskeletal re-organization [
120]. On the other hand, highly glycosylated ECM Lumincan promote colon cancer cells migration through binding with cell surface integrins, and activating actin cytoskeleton remodeling [
121,
122].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15010266