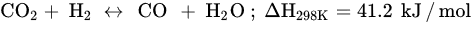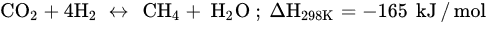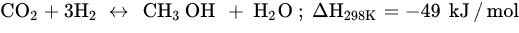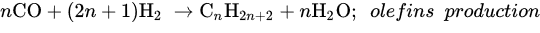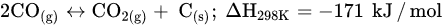Similar to other CO
2 hydrogenation reactions, methanation faces challenges, such as catalyst deactivation due to carbon deposition (fouling, Equation (5)) and decreased activity, resulting in a short catalyst lifetime. In order to prolong the catalyst lifespan and prevent catalyst degradation, CO
2 methanation catalysis has received huge attention to develop high-activity catalysts at low temperatures [
5]. Gao et al. designed an adequate ratio of CO
2/H
2 and found that a sufficient amount of H
2 can significantly influence the production of water vapor (H
2O), increase methanation activity rather than CO formation, and inhibit carbon deposition.
Various catalyst preparation methods, including conventional impregnation, sol–gel, and coprecipitation methods, have been developed to discover ways to achieve high activity and selectivity under mild conditions, good stability, and service life of CO2 methanation catalysts. Therefore, suitable active metals, such as Cu, Ni, Fe, Pd, and Co, have been reported for CO2 methanation, and Ni-based catalysts have been selected for industrial commercialization. However, these conventional metal-based catalysts often show low reaction activity at low temperature (180–300 °C) and tend to form particle agglomeration and sintering at high temperatures. For instance, Ni/Al2O3 catalysts prepared by wetness impregnation exhibit low metal dispersion and sinter in the presence of water at high temperatures, thereby increasing CO selectivity and energy consumption. Thus, active Ni metal with low loading is weakly dispersed and becomes less active at low temperatures, whereas the catalyst deactivates at high temperatures due to sintering. Catalyst design has been improved by adding promoters (Fe, Pd, Rh), additives, and support metal oxides (SiO2, ZrO2, CeO2) to enhance activity at low temperatures. However, addition of these components in the conventional method is detrimental toward efficient CO2 adsorption.
Core-Shell Nanostructured Catalysts for CO2 Methanation
Core-shell confinement of structure has garnered interest as a potential nanostructure support for CO
2 methanation. The activity and selectivity of core-shell structured catalysts are mainly affected by the type of metal used. Among porous shell supports [
13], metal-organic frameworks (MOFs) are a potential porous shell for Ni confinement because of their porous crystalline materials, high specific surface area, and tunable uniform elemental distribution. Ni precursors were treated solvothermally in a solvent solution at 136 °C to induce crystallization, and the Ni-MOF-74 suspension is calcined and then reduced in 5% hydrogen to form the Ni
xFe@C catalyst. The obtained catalyst achieves a CO
2 conversion of 72.3% with 99.3% CH
4 selectivity at 350 °C. Encapsulation of Ni-Fe alloy within carbon porous structure facilitates high CO
2 adsorption and effectively prevents the aggregation of active metal NPs during the reaction, thereby conferring the core-shell catalyst with superior stability. Moreover, the homogeneity of Ni-Fe NP elemental distribution can be preserved, which improves Ni dispersion.
New core-shell nanostructure based on cobalt (Co) catalysts have been successfully fabricated by Cui et al. [
78] to study catalytic performance of low temperature methanation. MnO-heterostructured NPs injected into porous graphitic carbon (Co/MnO@PGC) were synthesized via a single-step pyrolysis of bimetal CoMn@MOF-74. The resulting nanocomposite features an enriched Co/MnO heterointerface and exhibits excellent catalytic performance for low-temperature CO
2 methanation. The synthesized Co/MnO@PGC catalyst allowed CO
2 molecules to activate faster at a low heat of 160 °C over 99% selectivity with high STY
CH4 of 0.14 μmolCH
4⋅s
−1 gcat
−1. As the temperature reached 240 °C, CO
2 conversion and space–time yield (STY
CH4) rose to 32.1% and 13.34 μmol
CH4⋅gcat
−1⋅s
−1, respectively. At a high pressure (30 bar), STY
CH4 can reach up to 5.60 μmol
CH4⋅s
−1⋅gcat
−1 at 160 °C, which is even comparable to that of the optimal level of Ru-based catalysts. These results indicate that the synergistic interactions between Co and MnO NPs at the Co-MnO heterointerface are responsible for enhancing the catalytic activity toward CH
4 production at a low temperature. In addition, the Co/MnO heterostructured NPs encapsulated into PGC play an important role in preventing metal particle aggregation and improving thermal stability. High TOF
CH4 suggested that the Co/MnO heterointerface formed inside the PGC of Co/MnO@PGC can significantly boost its activity of low-temperature (160–220 °C) CO
2 methanation.
Core-shell metal@metal oxide particles can be promising as high thermal-conducting support materials owing to the high thermal conductivity of metal and excellent surface structural properties of the metal oxide itself. Various supports, such as Al
2O
3 [
80], CeO
2 [
81], ZrO
2, SiO
2 [
60], or zeolites [
60] have been proposed as metal oxide shell to protect the active metals at the core. In addition, Le et al. [
82] synthesized a Ni/Al@Al
2O
3 CSN catalyst for CO and CO
2 methanation by using hydrothermal surface oxidation (HTSO). Ni/Al@Al
2O
3 has selectively yielded carbonate and formate species, which suppress the CO intermediate. The confinement effect helped the CSN catalyst lower the activation energy barriers (74 kJ/mol), which outperforms the activation energy of conventional catalysts, namely, Ni/Al
2O
3 (80 kJ/mol) and Ni/SiO
2 (89 kJ/mol). Apparently, Ni/Al@Al
2O
3 CSN can successfully enhance the catalytic CO
2 adsorption owing to its high Ni dispersion and strong CO
2 binding.
Meanwhile, Ilsemann et al. [
60] prepared Co@SiO
2 and Co@Silicalite-1 catalysts via a solvothermal method to encapsulate the Co NPs inside two mesoporous structures of silica shells. They found that Co@SiO
2 improves the catalytic activity in low-temperature CO
2 methanation (230°C–400 °C) by suppressing the side reaction (RWGS), which results in highly selective CO
2 hydrogenation to methane. The thermal stability provided by mesoporous silica could preserve the active Co metals at elevated temperatures. However, CO methanation causes slight coking, resulting in a shift of kinetic stability and reduction in methane yield. Similarly, a silicalite-1-confined Ni catalyst was prepared through the selective desilication of the molecular sieve to produce extra voids and pore channels to cage Ni in the crystal [
83]. The Ni@Silicalite-1 catalyst is characterized by higher CO
2 conversion and CH
4 selectivity than conventional Ni/Silicalite, which can be attributed to the higher Ni fine dispersion in the void of silicate. The catalyst maintains stable performance over 50 h at 450 °C.
CSNs are expected to provide an anti-sintering effect by anchoring the active metal NPs in the porous channel. The formation of metal–metal oxide compounds modulates strong interaction to realize the low deactivation rate of reaction. However, this core-shell design has some shortcomings, such as complex preparation, expensive instrumentation, and lack of exposed defect surface, all of which restrict its industrial applications. Therefore, Yang et al. [
81] prepared Ni-phyllosilicate@CeO
2 CSN by using a hydrothermal method, to create Ni fine dispersion (3.3–6.3 nm). The anchored Ni phyllosilicate could further increase the H
2 and CO
2 uptake, contributing to high CO
2 conversion rate (65%), thus exhibiting high catalytic activity and stability for 100 h lifetime CO
2 methanation.
Le et al. [
84] continued their work on the Ni/Al@M-Al
2O
4 core-shell catalyst by promoting various transition metals (M = Mg, Ni, Co, Zn, or Mn) to develop a synergistic interaction between M/Al and enhance the catalytic activity of the CSN in low-temperature CO
2 methanation. Different influences of thermal conductivity on shell NP dispersion were observed as an anti-sintering property. Ni/Al@M-Al
2O4 CSNs were prepared using deposition-precipitation (DP) and wet impregnation (WI) methods to facilitate superior heat conductivity and surface properties for highly exothermic and endothermic reactions and control the effect of parameters on metal particle size. Morphological analyses showed that 9 wt% Ni/Al@MnAl2O4 (DP) has a significant BET surface area of 129 m
2/g and the highest Ni metal dispersion (9.7%) among other synthesized catalysts. Introduction of Mg into the spinel Ni/Al@MgAl
2O
4 CSN has provided a larger BET surface area of 171 m
2/g with the same dispersion quality to Ni/Al@MnAl
2O
4. Ni/Al@MnAl
2O
4 (DP) and Ni/Al@MgAl
2O
4 (DP) have demonstrated better catalytic performance than the catalyst prepared using the WI method. Both catalysts selectively produce high methane yield by hindering more chain of hydrocarbon while facilitating 90% CO
2 conversion under 300 °C, thereby promoting high catalytic activity for CO
2 methanation. Bimetallic Ni-Al has facilitated functional heat transfer across Ni/Al@M-Al
2O
4 CSN catalyst particulates as the Al metal releases high heat conductivity. Interestingly, Ni/Al@MnAl
2O
4 shows superior catalytic stability because it has a lifetime of 50 h while preventing coke deposition and Ni particle agglomeration during CO
2 methanation.
Considering the sharp rise in temperature of methanation and rapid catalyst deactivation by Ni particles, Wang et al. [
85] suggested the interaction of Mg with Ni as a bimetallic core in a Ni/Mg@MCM-41 duo-core@shell catalyst prepared using an in situ hydrothermal method with different Mg contents. Wang et al. also synthesized a conventional Ni/MCM-41 by using WI for comparison of catalytic performance. They found that Ni/MCM-41 features a larger BET surface area of 622.5 m
2/g than 0.05 wt% Ni/Mg@MCM-41, and the specific surface area continues to decrease as an additional 0.05 wt% Ni is incorporated. This result can be ascribed to the blockage of Mg particles on the pores when Mg
2+ has less tendency to replace Si
4+ ions in the SiO
2 lattice. However, the Ni/Mg@MCM41 core-shell catalyst produces better atomic composition than the conventional Ni/MCM41 catalyst. Under an optimal temperature of 360 °C, 0.05 wt% Ni/Mg@MCM-41 exhibits the highest CO
2 conversion at 88% during CO
2 methanation. The selectivity of CH
4 gradually decreases with increasing temperature, proving that high temperatures are not advantageous to CH
4 production because CO
2 methanation is an exothermic reaction. Regardless, the 0.05 wt% Ni/Mg@MCM-41 catalyst shows high catalytic activity at low temperatures (below 360 °C) and a large specific surface area (606.3 m
2/g), which are suitable for CO
2 methanation.
A well-defined nanostructured Ni@SiO
2 core-shell catalyst (diameter size of 27.1 nm) was synthesized with distinct metal–metal oxide interfaces in proximity to each other to carry out CO
2 hydrogenation [
86]. Noteworthy, the Ni@SiO
2 interface in the catalyst is responsible for RWGS reaction to form CO selectively. The strong interaction between Ni core and SiO
2 shell effectively restrains NP growth (agglomeration) and carbon deposition. Thus, the Ni@SiO
2 core-shell catalyst, yields 89.8% CH
4 and successfully converts 99.0% of CO molecules. Moreover, it retains high catalytic stability in CO methanation under a 100 h lifetime condition, which surpasses the stability of the conventional Ni/SiO
2 catalyst, whose CO conversion collapses after 12 h lifetime.
Ni@mpCeO
2 CSN was synthesized using nanocasting, followed by strong electrostatic adsorption for CO
2 methanation [
74]. The turn of frequency (TOF) for the Ni/mpCeO
2 CSN catalyst (0.183 s
−1) at 225 °C is threefold higher than that of the Ni catalyst supported on conventional CeO
2 prepared using the same method. Compared with the Ni catalyst, the Ni/mpCeO
2 CSN provides a rich NiCeO
2 interface with more oxygen vacancies, playing a key role in CO
2 activation. CO
2 activation over the Ni/mpCeO
2 CSN catalyst occurs through combined associative and dissociative mechanisms that have been observed through DRIFT mechanism study. Ni NPs are highly dispersed in the channels of mpCeO
2, which enhance H
2 dissociation, thereby supplying sufficient *H species for the formation of CO and *HCO intermediate species owing to high CH
4 selectivity. In addition to enhanced low-temperature activity and selectivity, the Ni/mpCeO
2 catalyst maintains its stability 70 h on stream because Ni sintering has been suppressed by the confinement effect of mesoporous CeO
2 structure. This study demonstrates the importance of the Ni-CeO
2 interface, at which high oxygen vacancy concentration facilitates CO
2 adsorption and activation while the adjacent Ni active sites accelerate H
2 dissociation.
2.2. CO2 Hydrogenation to Methanol
Among all possible products in the CO
2 hydrogenation reaction mentioned in the previous section, methanol (MeOH) is the most attractive (Equation (3)). MeOH is a clean, biodegradable, high-energy fuel and is highly versatile as it can easily generate other valuable fuels, such as DME, olefins, hydrocarbons, and long-chain alcohols [
27,
87,
88]. Furthermore, combustion of MeOH generates few carbon side products owing to its freezing point (−96 °C). Thus, MeOH is suitable as a hydrogen carrier without producing a huge number of SO
x or NO
x.
Unlike methanation, MeOH synthesis has thermodynamic and kinetic limitations, such as its high-pressure requirement for complete CO
2 activation and low reaction temperature to selectively enhance the methanol yield [
46,
89] Nevertheless, this reaction still produces a competing side reaction, RWGS. Therefore, a potential route to suppress CO production must be discovered to maintain high methanol selectivity. CO
2 hydrogenation to MeOH is favored at high pressures. Therefore, stable catalysts that are resilient to high temperatures and pressures are required. Active transition metals (Ni, Ru, Ga, Cu, and Co) are usually used for CO
2 hydrogenation to MeOH because of their high activity at certain temperatures, abundance, low cost, and different oxidation states/phases to improve catalytic stability and selectivity. However, such active metals can encounter rapid deactivation (sintering, fouling, and poisoning) because methanol synthesis is a naturally structure-sensitive reaction, thus limiting CO
2 activation.
Cu-based thermal catalysts are those used most often and hold many advantages for effective commercialization at the industrial scale. Cu catalysts are strongly active toward high methanol selectivity because of their three oxidation states (Cu
0, Cu
1+, and Cu
2+) [
90,
91,
92]. Cu provides active sites for H
2 dissociation, and metal oxides increase the number of active sites for CO
2 activation. Wang et al. reported that methanol selectivity corresponds to the proportion of strong basic sites to the total basic sites. Despite their benefits, Cu NPs agglomerate into large particles at elevated temperatures, which decrease MeOH yield. Therefore, understanding the changes in different surface atom arrangements on Cu NPs is important because the homogeneity of the catalyst structure affects the catalytic activity.
Core-Shell Nanostructured Catalyst for CO2 Hydrogenation to Methanol
CSN catalysts are attractive for these conversions because of the impact of the shell materials on reaction selectivity and catalyst stability [
93] The core-shell nanostructure and surface have many advantages, such as enhancement of the essential properties of conventional catalysts. For example, commercialized Cu/ZnO/Al
2O
3 catalysts are unstable under high partial pressure at elevated temperatures, accelerating steam in the reaction atmosphere, hence causing rapid agglomeration and undesired crystal growth [
3,
27,
94,
95] A core-shell arrangement may optimize the interaction between the metal and porous support and minimize Cu sintering by creating unique multifunctionalities of catalytic sites.
An et al. [
96] investigated Cu NPs coated with Zn to boost the surface electronic concentration and surface adsorption from high bimetallic synergy. The Cu/Zn bimetallic particles were anchored within the MOF network, and CuZn@UiO-bpy CSN was suggested to enhance the active metal dispersion and the SMSI. Cu NPs form within a diameter of 0.5–2 nm, depicting that the NPs are homogeneously dispersed, thus spreading more active sites and catalytic activity. Such structural properties result in remarkable CO
2 conversion (17.4%) and methanol selectivity (85.6%). In addition, CuZnO@UiO-bpy CSN shows three times higher methanol yield at 250 °C than the conventional catalyst. This report agrees with the findings of Tisseraud et al.’s compilation study [
97,
98,
99] where Cu@ZnO
x CSN catalysts exhibit 100% methanol selectivity as the oxygen deficiency formed by Zn migration provides active sites and hinders CO formation by side reactions (RWGS or MeOH decomposition).
A recent study has prepared a CuIn@mSiO
2 core-shell catalyst using a two-step solvothermal synthesis [
57]. This catalyst was compared internally with Cu@SiO
2, In@SiO
2, and conventionally prepared CuIn/SiO
2 catalysts. A perfect core-shell shape has been successfully synthesized. The activity results showed that CuIn@SiO
2 outperforms the others in terms of CO
2 conversion but exhibits the second lowest methanol selectivity (21.8%) owing to CO formation at 250 °C. Nevertheless, the most stable performance over 100 h is dominated by Cu@SiO
2 with zero sign of carbon deposits. Considering that mesoporous silica (mSiO
2) shells are highly effective in limiting metal agglomeration and preserving the original metal particle sizes by providing a layer of thermally stable surface, Yang et al. [
100] embedded Cu/ZnO within a layer of mSiO
2 and achieved stable CO
2 conversion and methanol yield over 160 h lifetime at a low temperature (260 °C). They observed that Cu/ZnO@mSiO
2 shows better catalytic performance than the conventional impregnated catalyst Cu/ZnO/SiO
2, which is deactivated after only 20 h on stream.
Apart from tuning the product selectivity and preventing active sites sintering, the core-shell catalyst can help fix and activate CO
2 molecular activation. Hydroxyl species, which can be obtained from transition metal phyllosilicate (TM@SiO
2p), enhance CO
2 hydrogenation. Jangam et al. [
94] prepared Cu-SiO
2p via a hydrothermal method and compared its performance with conventionally impregnated Cu-SiO
2 for CO
2 hydrogenation to MeOH at 200–350 °C. Cu-SiO
2p reduced at 225 °C produces a stable CO
2 conversion and methanol selectivity of 3.5% and 77%, respectively.
Recently, hollow Cu@ZrO
2 derived from a MOF network has been developed through pyrolysis for selective CO
2 hydrogenation to methanol [
101]. The hollow structure provides easy access of CO
2 and H
2 to diffuse on active sites. Han et al. found that the basic sites of Cu-ZrO
2 interfaces are responsible for the main adsorption and activation sites of CO
2. The core-shell confinement structure yields a high methanol selectivity of 85% at 220 °C.
Other than Cu-based catalysts, noble metals, such as Pd-based heterogeneous catalysts also received recognition for CO
2 hydrogenation. The electronic structure of Pd-based catalysts plays significant roles in the reaction because its metallic sites can be tuned to obtain high-stability catalysts. However, their applications for large-scale plants are limited by sintering and expensive source. Xiao et al. [
102] have recently designed stale Pd NPs in a confined environment. Pd@Cu core-shell was confined within a layered double hydroxide through modified coprecipitation. They conducted a catalytic test on formate species formation to identify the feasible CO
2 hydrogenation pathway without forming CO intermediates. Pd
0.4@CuMgAlO
x with a CO
2/H
2 composition of 20:20 successfully yielded 5.68 mmol∙g1∙h-1 of formate, whereas the core-shell catalyst showed no significant loss in formate yield after the fourth cycle, confirming its excellent stability. Kinetically, the Pd metallic sites govern the H
2 dissociation by forming active Pd-H.
3. CO2-Reforming Reactions
Although renewable H2 production via electrolytic, photoelectrochemical (PEC), and solar thermochemical methods is promising, cost effective, abundant, and sustainable, it is still not viable for industrial commercialization. Thus, thermocatalytic hydrogen production, which is a nonrenewable route that is effective for rich H2 production from biomass, is still relevant in the gas production market. The reforming of fossil fuels, especially natural gasses, via decomposition of hydrocarbon molecules to release H2 is the most common source for H2 production globally. Traditionally, H2 can be produced via several processes, such as steam methane reforming, partial oxidation reforming, methane pyrolysis, coal gasification, and DRM.
3.1. CO2 Dry Reforming of Methane
Unlike the abovementioned methods, DRM offers low operating cost, utilizes two hazard greenhouse gases (CO
2 and CH
4) to produce highly pure gas (CO/H
2), and allows easy processing of value-added hydrocarbons and chemicals via the Fischer–Tropsch process [
104]. DRM is a more economical process relative to other methods because it eases the gas separation of final products. CO
2 utilization has a significant influence on DRM performance, considering that the adsorption isotherm and its activation are the main steps to achieve optimal H
2 production.
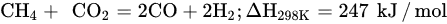
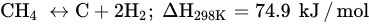
DRM is an endothermic reaction that requires excessive heating (>700 °C) driven by the following main reaction (Equation (6)). Poisoning, catalyst deactivation, and coke deposition are the common issues faced in DRM at high temperature because of methane decomposition (Equation (7)), Boudouard reaction (Equation (5)), and CO2 hydrogenation (Equation (1)), whereas the RGWS reaction usually occurs in DRM at temperatures below 800 °C. To limit the RWGS reaction, DRM must operate at high temperatures, approximately 900 °C, to achieve high yields of H2 and CO. Hence, key factors of efficient and feasible DRM reaction are optimized temperature, pressure, CH4/CO2 ratio, and catalyst design and composition.
An effective DRM reaction mechanism theoretically involves multidisciplinary transitional states, such as methane dissociative adsorption, CO
2 dissociative adsorption, hydroxyl group formation, and intermediate oxidation and desorption [
105]. In detail, CH
4 gases must dissociate on the catalyst surface sites to complete their tetravalency, whereby CH
3 molecules are adsorbed on top of active metal atoms while another CH
2 molecule occurs between two metal atoms, called step sites. Then, another greenhouse gas, CO
2, breaks its double bond, where C-O is adsorbed on the surface between the metal and support, leaving one oxygen atom exposed. Later, the H
2 molecules migrate from the metal particles to the support atoms to form hydroxyl (
−OH) species at temperatures below 800 °C. Finally, the metal-surface oxygen provided by high oxygen mobility support reacts with the S-CH
x species to form new S-CH
xO intermediates, which potentially form as CO and H
2. This kinetic reaction of DRM is influenced by surface electronic properties.
Efforts have been exerted to develop novel catalysts that can increase CO
2 and CH
4 activities at low temperatures. Nickel is the most prevalent metal-based catalyst because of its abundancy and low cost, making it suitable for industrial catalytic processes; however, Ni is rapidly deactivated because of high carbon formation from side reactions, either methane cracking or CO disproportionation [
105,
106] Noble metals, such as Pd [
106], Ru, and Pt [
107,
108,
109,
110], are suggested to replace Ni because they are highly resistant to carbon formation. However, their expensiveness restricts their promising properties in the larger market. Hence, catalysts that minimize coke formation and preserve the active sites in DRM need to be developed.
Core-shell catalysts may offer high thermal stability, sintering resistance, and several functionalities, which aid in reducing the rate of carbon deposition [
111,
112]. The core-shell structure also promotes good control of dispersion and preservation from metal agglomeration, resulting in enhanced catalyst stability. Porous materials with high thermal resistance and optimum porous size channels that could anchor the active metal core are ideal to stabilize the metal NPs and minimize metal sintering at elevated temperatures [
113].
Core-Shell Nanostructured Catalyst for CO2 Reforming of Methane
Several studies on CSN catalysts for DRM have been published. Metals confined with mesoporous channels are the most extensively investigated catalysts in this field. Metal–support interaction is important in driving high activity of DRM; thus, a strong interface relationship is often demanded to inhibit active metal agglomeration [
114].
Ni NPs embedded into zeolitic materials, such as silicalite, have been widely accepted as a promising structure for Ni confinement, but over 20 wt% Ni loading decreases the dispersal and encapsulation. Recently, Liu et al. have developed a controlled “dissolution–fractional crystallization” method of confining high loading and uniform Ni NPs into the hollow silicalite-1 (S-1) shell [
115]. Active species of Ni as core has maintained its particle size to ca. 4–5 nm with optimized Ni loading from 3% to 20%, and the TOF remains at ca. 60 s
−1 (800 °C) in the dry reforming of methane reaction. The augmented density of active sites with Ni loading renders an outstanding reaction rate of 20.0 mol
CH4/g
cat/h over 20% Ni@S-1.
A multiple core-shell structure using indium–nickel (In-Ni) intermetallic alloy as core and SiO
2 as porous shell has been successfully synthesized using the Stober method [
116]. The InNi@SiO
2 CSN displays superior coking resistance for DRM reaction by minimizing the amount of carbon formation in DRM reaction. Even for the sample with only 0.5 wt% In doping, In
0.5Ni@SiO
2 CSN has been achieved based on the balance of coke deposition resistance and DRM reactivity. Hypothetically, lowering in loading confers coke resistance, whereas high In loading leads to low catalytic activity because of the formation of InNi
3C
0.5 species. Moreover, the specific surface area of the core-shell catalysts does not change in structural behavior even after reduction during the reaction for 20 h, indicating that no observed coke blocks the pore channels during the long-term thermal operation. The increase in electron-cloud density on Ni can weaken the ability of Ni to activate the C–H bond and decrease the deep cracking of methane. The binding energy of Ni
2p3/2 in the InxNi@SiO
2 catalyst decreases with increasing in loading, which means the interaction between Ni and Ni is weakened, and the interaction between Ni and In is enhanced, indicating that Ni particles are not easy to be sintered.
Lu et al. [
117] have developed a novel structure catalyst of Ni@S2-T with Ni NPs highly dispersed in silicalite-2 zeolite (S2) via a two-step method involving the microemulsion method followed by solvent-free crystallization. S2 is a silica analog of aluminosilicate zeolite (ZSM-11), which consists of four- to six-member rings chained to create a porous channel system with 10-member ring openings. The unique pore structure and channel of S2 lattice can improve the confinement efficiency by strengthening the metal–support interaction caused by the formation of Ni phyllosilicate intermediate in the shell, which is regarded as the main reason for the superb catalytic performance of Ni@S2-T for DRM. Compared with Ni-SiO
2 prepared by microemulsion, Ni/S2 by impregnation, and Ni@S2-O by direct crystallization, Ni@S2-T catalyst exhibits optimal catalytic activity and stability for DRM. The catalyst achieves long-term stability, over 100 h, as the conversion of CH
4 maintains its value of 25%. Furthermore, after the Ni@S2-T catalyst is overspent, hardly any coke can be found after the prolonged test, which indicates the remarkable anti-coking ability of Ni@S2-T.
Kong et al. controlled the porous size of SiO
2 channel under intrinsic hydrothermal treatment to allow precise control over the Ni surface [
118]. The synthesized Ni@SiO
2 CSNs show that the confinement structure can destroy large Ni ensembles and form metastable Ni−O⋅Si centers. CH
4 activates on the small fraction of Ni to form CHx rather than carbon. Moreover, the Ni−O⋅Si center stabilized by interfacial confinement provides labile oxygen to oxidize CH
x. This Ni catalyst exhibits highly stable activity under 800 °C, CH
4: CO
2 = 2:1, and 5 bars without carbon deposition for 100 h, where carbon formation is thermodynamically much favorable.
Another breakthrough of core-shell structures defined as hollow nanostructures has been intensely researched for their delimited cavity and enclosed shell [
119], which could manifest tunable focal properties aside from well-defined active sites, thus enhancing the catalytic functionality. Herein, Kosari et al. modified Ni-SiO
2 hollow spheres (HSs) with different shell thicknesses and interior cavity sizes via a hydrothermal method [
120]. As a result, final hollow Ni-SiO
2 exhibits varied shell thicknesses from 11 nm up to double-shell morphology with 77 nm being the inner shell distance. NiHS-SiO
2-S renders CH
4 conversion equal to 69%, which is higher than those of other low Ni-loaded catalysts. However, DRM activity rate decreases as the shell thickness of SiO
2-derived samples is increased further. Interestingly, no coke formation during the reactivity tests indicates that the formulated NiHS-SiO
2 catalyst is a promising candidate to catalyze the DRM reaction while acting as a strong carbon resistance material.
Recently, Marinho et al. have developed a core-shell confined structure, a Ni-based mesoporous mixed CeO
2-Al
2O
3 oxide catalyst by top–down synthesis, and an evaporation-induced self-assembly (EISA) method to overcome Ni particle sintering in high-temperature DRM reaction [
80]. EISA utilizes mesoporous bimetallic oxides to control the shape and robustness of the Ni@CeO
2/Al
2O
3 catalyst by presenting as mesoporous structures with highly dispersed Ni in the form of NiAl
2O
4 spinel clusters. Small (<5 nm) and homogeneous metallic Ni particles form after reduction steps. In addition, the Ce species in the structure reinforce the strong metal–support interaction with Al
2O
3, which enhances oxygen mobility and acts as spatial sites for CO
2 adsorption, thereby increasing the catalytic activity and promoting the carbon removal mechanism. Therefore, Ni@CeO
2-Al
2O
3 catalysts prepared by one-pot EISA exhibit high activity and stability for DRM owing to the successful encapsulation of Ni particles and coke resistance.
Wang et al. [
121] prepared basic metal oxide (MgO and La
2O
3)-modified Ni confined in dendritic mesoporous silica catalysts (Ni-MgO@DMS and Ni-La
2O
3@DMS) via a sol–gel method. The Ni-MgO@DMS CSN formed exhibits a completely confined structure and yields optimum conversion rates of CH
4 and CO
2 up to 35% and 40%, respectively, in a low-temperature DRM reaction of 550 °C. Remarkably, only a few insignificant carbon depositions are observed during the 8 h time-on-stream stability evaluation, which can be attributed to the fast alkaline oxides adsorption and CO
2 activation. Moreover, the modified Ni-MgO@DMS and Ni-La
2O
3@DMS CSNs show high Ni sintering and carbon resistance owing to the high oxygen vacancy facilitated by the presence of magnetic oxide species Mg and La, contributing to its hydrocarbon species (CHx) activation. As the reaction time is extended to 50 h, the spent Ni-La
2O
3@DMS catalyst has a negligible amount of carbon formation.