Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Andrology
Ferroptosis, an iron-dependent type of regulated cell death, is triggered by the accumulation of lethal lipid peroxides. The cellular iron metabolism is essential for maintaining life activities in normal biological processes, including the generation of cellular energy, oxygen transport, heme synthesis, DNA synthesis and repair, etc. Notably, studies show that the iron metabolism is also quite important for the reproductive system and is intimately related to spermatogenesis. The relationship between the abnormal iron metabolism and ferroptosis in male reproductive disorders is gaining increasing attention.
- ferroptosis
- iron metabolism
- GPX4
- lipid peroxidation
- male reproduction
1. Cellular Iron Metabolism
The mammalian iron metabolism could be divided into four sections: absorption, storage, utilization, and efflux. The main mechanism for the intake of extracellular iron is a transferrin-bound iron (TBI) import, which relies on the iron transporter transferrin (Tf) and the transmembrane glycoprotein Tf receptors (TfR). The other mechanism is the non-transferrin-bound iron (NTBI) input, which depends on the cellular membrane protein represented by SLC39A14(ZIP14) [15]. First, each Tf is combined with two extracellular ferric ions (Fe3+), which subsequently bind to the TfR in TBI. Then, the Tf-TfR complex is internalized into the endosome. As the environment in the endosome is acidic, ferric ions are released from the complex, while Tf and TfR will be circled to the cell surface for the next round of iron absorption [16]. Next, the ferric ions are rapidly reduced to ferrous ions (Fe2+) by STEAP3 metalloreductase. Following this, Fe2+ would be exported to the cytoplasm from the endosome by a divalent metal transporter 1 (DMT1) and forms the labile iron pool (LIP). Iron in the LIP is loosely bound to negatively charged small molecules and proteins [17], so its catalytic activity is difficult to control. Notably, LIP is the transportation hub that links the iron metabolism mentioned above. The iron could exit the LIP in one of the four main pathways: (i) the majority of iron is transported to ferritin, which is a ubiquitous iron-storage protein in the cytoplasm and can combine up to 4500 iron atoms [18]. (ii) Iron can also be utilized directly in the cytoplasm as a cofactor of cytoplasmic proteins. (iii) Iron could be trafficked to the mitochondria via SLC25A37 and SLC25A28 to participate in heme and iron-sulfur (Fe-S) cluster synthesis [19] or to be stored in mitochondrial ferritin (MtFt) [20]. However, heme could be decomposed into Fe2+ under the catalysis of heme oxygenase 1(HO-1), which increases the LIP in turn [21,22]. It is worth noting that mitochondrial iron is crucial for spermatogenesis. Research revealed that the number and vitality of sperm were reduced in mitoferrin-2 knockout mice [23]. Furthermore, the down-regulation of the mitoferrin gene resulted in germ cell developmental defects in Drosophila, some of whose mitochondrial iron metabolism genes are similar to mammals in their expression patterns [24]. (iv) Excess cellular iron in the LIP would be released outside the cell by ferroportin (FPN), which is the only known cellular iron exporter (Figure 1).
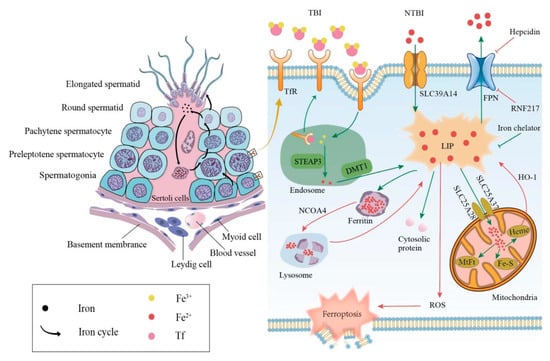
Figure 1. Relationship between iron metabolism and ferroptosis during spermatogenesis. The left side shows the iron circle in the process of normal spermatogenesis. The right side shows cellular iron metabolism and results caused by iron overload. TBI, transferrin-bound iron; NTBI, non-transferrin-bound iron; Tf, transferrin; TfR, transferrin receptor; DMT1, divalent metal transporter 1; FPN, ferroportin; LIP, labile iron pool; MtFt, mitochondrial ferritin; HO-1, heme oxygenase 1; NCOA4, nuclear receptor coactivator 4; ROS, reactive oxygen species.
In addition to the general iron metabolism, there is a largely unique autonomous iron cycle in spermatogenesis (Figure 1). Iron is taken up by spermatogonia and preleptotene spermatocytes via TBI and continuously transferred to round spermatids with their self-renewal. In particular, a proportion of iron is released by round spermatids and cycled to the apical compartment of the Sertoli cells, with the rest carried away by elongated spermatids. Sertoli cells store the acquired iron in ferritin, which would be secreted and returned to primary spermatocytes. Although the elongated spermatids would take a part of the iron away from seminiferous epithelia, spermatogonia and preleptotene spermatocytes could absorb iron to compensate for the loss via the TBI mentioned above. This internal iron cycle protects the developing germ cells from peripheral iron fluctuations, thus ensuring the stability of spermatogenesis [25].
2. Abnormal Iron Metabolism Leads to Dysfunction of Male Reproductive System
The abnormal iron metabolism, either an iron excess or iron deficiency, would affect spermatogenesis and cause a reproductive system dysfunction. Here, we mainly discuss the damage caused by an iron overload in the male reproductive system. The principal causes of an iron overload in testicular cells are the degradation of the reservoir or obstruction of the outlet. Once ferritin is degraded through autophagy under the regulation of the autophagic receptor nuclear receptor coactivator 4 (NCOA4), the large number of iron atoms stored in it would be released to the LIP [26]. The degradation of FPN, whether mediated by acknowledged hepcidin [27] or newly discovered ubiquitin ligase RNF217 [28], hinders the iron output and increases the internal iron content as well. As mentioned above, the LIPs catalytic activity is unrestricted when iron-overloaded; thus, ferroptosis in testicular cells can be triggered in the following two ways. On the one hand, electrons can be transferred to hydrogen peroxide via the Fenton reaction, Fe2+ + H2O2→Fe3+ + OH− + •OH, forming the extremely damaging hydroxyl radical [29]. On the other hand, iron-dependent oxidases, including lipoxygenases, xanthine oxidases, NADPH oxidase, or catalase [30] could generate soluble and lipid ROS to promote ferroptosis (Figure 1).
Ferroptosis is a newly discovered cause of clinical diseases of the male reproductive system. A study showed that ferroptosis was involved in the occurrence of asthenozoospermia and led to an impaired sperm function. The iron levels in the semen of asthenozoospermic patients were significantly higher than those of the normal group, accompanied by an increase in ROS [8]. Furthermore, it is noteworthy that smokers have higher levels of ferroptosis than non-smokers among infertility patients. Heavy smokers have significantly more iron and ROS in seminal plasma and problems with sperm abnormalities, such as sperm vitality and progressive motility [6].
The down-regulation of FPN is particularly noticeable in animal and cell models, created for studying male reproductive system disorders. After an exposure to low-dose Cd, the expression of FPN was significantly down-regulated, with an elevated total iron and ferrous iron levels in testicular cells. This exposure also resulted in the massive loss and detachment of testicular germ cells, as well as a decreased meiotic index and testicular weight in mice [31]. However, the expression of the input proteins—not only TfR but also ZIP8 and ZIP14—was not altered. Interestingly, ZIP8 and ZIP14 played major roles in the uptake of NTBI, and the iron input in this way increased the potentially harmful LIP in liver cells [15,32]. Furthermore, Zhao et al. found that a busulfan exposure led to ferroptosis in mice testes, which was manifested by the down-regulation of FPN, the accumulation of iron, an increased malondialdehyde (MDA) content, and the morphological changes. They further confirmed that ferroptosis was caused by down-regulated FPN and GPX4. After treatment with deferoxamine (DFO), an iron chelator, the iron content and abnormal changes mentioned above were all reversed [5]. The decreased expression of the FPN protein and mRNA resulted in elevated iron and lipid ROS levels, and ferroptosis was inhibited by either a DFO treatment or FPN overexpression in ischemia–reperfusion-injured TM4 cells, which were the testicular Sertoli cells of normal mice [33]. A significant increase in intracellular iron and ferroptosis also occurred in the TM4 cells exposed to PM2.5, which are fine particles with a diameter of 2.5 μm or less [7].
Different testicular cells are differentially sensitive to iron accumulation-induced ferroptosis. Round spermatids showed iron-dependent cell death induced by 4-hydroxynonenal (4HNE), while pachytene spermatocytes could not be induced to ferroptosis by 4HNE [34]. In Leydig cells, tetramethyl bisphenol A (TMBPA) significantly increased the testicular iron content, induced ferroptosis, and inhibited the testosterone synthesis during late puberty [35]. An iron/fat-enriched diet led to the degeneration of seminiferous tubules in mice, an increase in vacuoles and space in the basement membrane, and a decrease in the total testosterone levels in the testis.
Contrary to the above, the decrease in the iron content in the LIP would inhibit ferroptosis [37], but it is still unfavorable to spermatogenesis. A lack of iron could lead to iron deficiency anemia (IDA), which induces an anoxic environment in the testis. An experimental article reported that after the correction of IDA, the patients’ testosterone levels and sperm parameters were significantly improved, especially in those who had subnormal values of these parameters [38]. Interestingly, the latest research shows that an iron supplementation could abrogate the testis dysfunction due to an iron deficiency through the regulation of the testicular antioxidant capacity [39].
In summary, the stabilization of the intracellular iron balance is essential to maintain the normal process of spermatogenesis. An iron overload leads to ferroptosis in germ cells, and inhibiting it, such as by using iron chelation or up-regulating the expression of FPN, is likely to provide an effective approach to reversing this type of death.
This entry is adapted from the peer-reviewed paper 10.3390/genes14010043
This entry is offline, you can click here to edit this entry!