Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Macrophages are effector cells of the innate immune system, which can play a crucial role in the generation of anti-tumor immunity through their ability to phagocytose cancer cells and present tumor antigens to the cells of adaptive immunity. However, the macrophages that are recruited to the tumor microenvironment predominantly play pro-tumorigenic roles. Several strategies targeting pro-tumorigenic functions and harnessing the anti-tumorigenic properties of macrophages have shown promising results in preclinical studies, and a few of them have also advanced to clinical trials.
- TAMs
- inflammation
- prognosis
1. Origin and Phenotypic Plasticity of TAMs
TAMs have long been believed to be originated from bone marrow-derived monocytic precursors which migrate into the TME by chemotaxis [16,17]. Recent studies indicate that tissue-specific embryonically derived macrophages also infiltrate tumor tissues and constitute a significant source of TAMs [8,16,17]. Macrophages are one of the most plastic immune cells, capable of acquiring distinct functional phenotypes depending on the microenvironment signals (Figure 1A). They can reversibly transform into anti-tumorigenic M1-like or pro-tumorigenic M2-like phenotypes in response to specific stimuli in the TME. The Th1 cytokines (IFNγ, TNF-α,) and bacterial-derived LPS promote the differentiation of monocytes into M1 phenotype. Whereas M2 differentiation occurs in response to Th2 cytokines (IL-4, IL-10, TGF-β1) and PGE2 produced in the TME [8,10,16]. The M1 and M2 division of TAMs is based on the surface markers expressed by these two types of macrophages. HLA-DR, CD80, CD86, CD197, TLR-2,4, and iNOS are dominant surface markers of M1 macrophages, whereas the M2 macrophage express CD163, CD209, CD206, FIZZ1, Ym1/2, and CCL2 are on their surface. The M1 macrophages retain their intrinsic ability to phagocytose and produce anti-tumor inflammatory responses [10]. The efficient tumor antigen-presenting ability of M1 macrophages promotes recruitment and stimulation of other leukocytes to exert their cytotoxic functions against cancer cells. For example, the immunostimulatory cytokines (IL-6, IL-12, TNF) released by M1 macrophages can enhance the activity of CD8 + T-cells and NK cells (Figure 1A). On the other hand, M2-type macrophages are endowed with a variety of tumor-promoting capabilities including immunosuppression, angiogenesis, neovascularization, as well as the activation of stromal cells [8,10,16,17,18]. However, it is worth mentioning that although TAMs are often considered identical to M2-type macrophages, a transcriptomic analysis of TAMs isolated from a murine fibrosarcoma revealed an unexpected transcriptomic profile of TAMs that differed from both M1 and M2 macrophages. In addition, an individual macrophage (TAM) can co-express markers of M1 and M2 phenotypes [19]. TAM diversity in tumors, therefore, is much more complex than the overly simplistic classification of TAMs into M1- and M2-type [20].
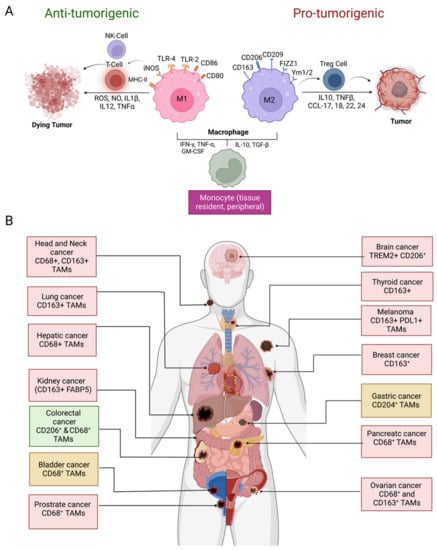
Figure 1. Macrophage polarization and prognostic significance in cancer. (A) Differentiation of monocytes in M1 and M2 macrophages in the TME and their role in tumor suppression and promotion, respectively; (B) prognostic significance of TAM density in various cancer types and IHC markers used for TAM identification. Presence of TAMs correlates with the unfavorable disease prognosis in glioblastoma, thyroid, lung, hepatic, kidney, ovarian, head and neck, breast, prostate, and melanoma (red boxes) whereas favorable prognosis (green box) in colorectal cancer. Both favorable and unfavorable correlations have been reported in gastric cancer and bladder cancer (yellow boxes). Abbreviations: NK, natural killer; ROS, reactive oxygen species; NO, nitric oxide; IL, interleukin; TNF, tumor necrosis factor; MHC, major histocompatibility complex; iNOS; inducible nitric oxide synthase; TLR, toll-like receptor, IFN, interferon; TGF, tumor growth factor; GM-CSF, colony-stimulating factor; Treg, regulatory T lymphocytes; FIZZ1, resistin-like molecule α, Ym1/2; chitinase-like protein Ym1/2, CCL; C-C chemokine ligand; CD, cluster of differentiation; FABP5, fatty acid binding protein 5; TREM, triggering receptor expressed on myeloid cells; PDL1, programmed death ligand 1 (Figure created with BioRender.com accessed on 15 December 2022).
2. Therapeutic Targeting of Macrophages
Several macrophage-targeting strategies are currently in the preclinical stages or are being tested in clinical trials. The focus of these strategies has primarily been on depleting tumor-associated macrophages, preventing monocyte recruitment to the tumor, inhibiting macrophage polarization towards the M2 phenotype, or re-educating polarized macrophage so that they can perform anti-tumor functions. Additionally, another focus area of therapeutic targeting using TAMs focuses on blocking inhibitory immune checkpoints, in particular the inhibition of anti-phagocytic immune checkpoints to enhance the phagocytosis of malignant cells. In addition to this, macrophages are also being harnessed as vehicles for delivering cytokines to achieve therapeutic responses and are also being developed as adoptive cell therapies to treat solid tumors (Figure 2). Various approaches to therapeutic targeting and harnessing macrophages have been discussed in this section. A timeline of milestones achieved in macrophage research and their therapeutic targeting is shown in Figure 3.
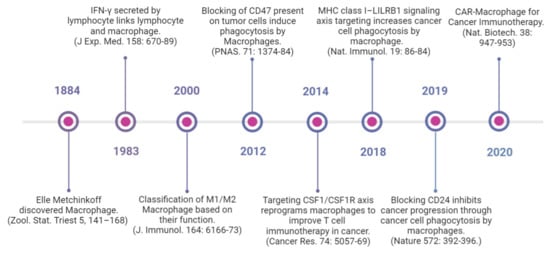
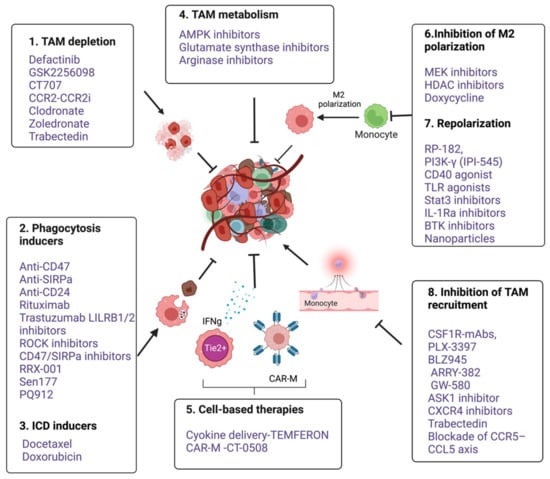
Figure 3. An overview of various anti-cancer immunotherapeutic modalities targeting tumor associated macrophages. 1. TAM depletion: Bisphosphonates such as clodronate-liposome [152], Zoledronate [15] and cytotoxic drug Trabectedin [153] and FAK inhibitors such as Defactinib, GSK2256098 and CT-707 (Conteltinib) [154,155], 2. Phagocytic inducers; Monoclonal antibodies targeting myeloid checkpoints such as CD47 and SIRPα in lymphomas, Anti-CD24 in ovarian and breast cancer [14], Rituximab targeting CD20 in B cell lymphoma [156,157], Trastuzumab targeting HER2 in breast and ovarian cancers [158,159]. LILRB1/2 inhibitors [160]. Small molecule inhibitors targeting CD47- SIRPα axis such as RRX-001 [161], Sen177 and PQ912 [162], Rho-kinase inhibitor (Y27632) in solid cancer models [163]. 3. ICD inducers; docetaxel, doxorubicin [164,165,166]. 4. Targeting TAM metabolism; Metformin, an AMPK inhibitor, 2-Deoxy-d-glucose (2DG), a Hexokinase-2 inhibitor [167]; Glutamine Synthetase inhibitor, Methionine Sulfoximine and arginase inhibitors (CB-1158 and L-Norvaline). 5. Cell based therapies- Macrophages as delivery vehicle; TEMFERON, TME delivery of IFNγ [168], and IFNα [169], CAR macrophages; CT-0508, targeting solid tumors [151]; 6. Inhibition of M2 polarization. Puerarin [170]. Trichostatin-A [171] and TMP195 [172], Doxycycline [173]. Stat-3 inhibitors alone and in combination with ERK inhibitor [174]. 7. Repolarization from M2 to M1; RP-182, a synthetic peptide [175]. Duvelisib (IPI-145), an oral inhibitor of the PI3Kδ and PI3Kδγ isoforms [176,177]. CD40 agonistic mAbs [178]. TLR7,8,9 agonist [179]. Ibrutinib, a BTK inhibitor [180]. Nanoparticles based delivery of TLR agonists, Bisphosphonates, DNA, mRNA, and miRNA [181]. 8. Inhibition of TAM recruitment; Antagonists of CSF1R, CXCR4, CCR5-CCL5 and CCL2-CCR2 (Table 1). GS444217, an ASK1(MAP3K5) inhibitor [182] (Figure created with BioRender.com accessed on 15 December 2022).
Table 1. Selected clinical trials of TAM-targeting drugs.
| Targeting TAMs Strategies | Name | Targets | Cancer Types | Phases | Clin. Trial |
|---|---|---|---|---|---|
| Phagocytosis | RRX-001 | CD47, SIRPα | Non-small cell lung cancer | Phase III | NCT03699956 |
| Hu5F9-G4 | CD47 | Advanced tumors | Phase I | NCT02216409 | |
| JTX8064 | LILRB2 | Advanced refractory solid tumors | Phase I | NCT04669899 | |
| Depletion of M2-like TAMs | Zoledronate | NA | Mammary carcinoma | Phase III | NCT00320710 |
| TAMs recruitment | Pexidatintinib | CSF-1R | Advanced solid tumors | Phase III | NCT02371369 |
| D2923 | CSF-1R | Myelogenous leukemia | Phase II | NCT04989283 | |
| Emactuzumab | CSF-1R | Advanced solid tumors | Phase III | NCT05417789 | |
| 3D185 | CSF-1R | Colorectal cancer | Phase II | NCT05039892 | |
| PLX3397 (Pexidarnitib) |
CSF-1R | Melanoma | Phase II | NCT02071940 | |
| CSF-1R | Advanced solid tumors | Phase I | NCT02734433 | ||
| CSF-1R | PVNS or GCT-TS | Phase III | NCT02371369 | ||
| CSF-1R | Leukemia, sarcoma, or neurofibroma | Phase I/II | NCT02390752 | ||
| CSF-1R | Acute myeloid leukemia | Phase I/II | NCT01349049 | ||
| PLX7486 (Plexxikon) | CSF-1R | Phase I | NCT01804530 | ||
| DCC-3014 | CSF-1R CSF-1R |
Advanced-stage or metastatic solid tumors | Phase I | NCT03069469 | |
| ARRY-382 | Phase I | NCT01316822 | |||
| LY3022855 mAb (IMC-CS4) | CSF-1R | Solid tumors | Phase I | NCT02265536 | |
| Phase I | NCT01346358 | ||||
| AMG820 mAb | CSF-1R | Solid tumors | Phase I/II | NCT01444404 | |
| MLN1202 | CSF-1R | Bone metastasis | Phase I/II | NCT01015560 | |
| AMG820 mAb/Pembrolizumab | CSF-1R | Solid tumors | Phase I/II | NCT02713529 | |
| BLZ945/PRD001 | CSF-1R | Advanced solid tumors | Phase I/II | NCT02829723 | |
| Cabiralizumab/Nivolumab | CSF-1R | Advanced solid tumors | Phase I | NCT02526017 | |
| MLN1202 | CSF-1R | Bone metastasis | Phase I/II | NCT01015560 | |
| RO5509554/RG7155 (Emactuzumab)/Paclitaxel | CSF-1R | Advanced solid tumors | Phase I | NCT01494688 | |
| PD-0360324 mAb/Cyclophosphamide | CSF-1R | Ovarian cancer | Phase II | NCT02948101 | |
| Eribulin | CSF-1R | Metastatic breast cancer | Phase I/II | NCT01596751 | |
| PF-04136309 | CCR2 | Pancreatic cancer | Phase II | NCT02732938 | |
| CCX872 | CCR2 | Pancreatic cancer | Phase I | NCT02345408 | |
| CCR2i | CCR2 | Cutaneous t-cell lymphoma | Phase II | NCT02732938 | |
| mNOX-E36 | CCL2 | Glioblastoma | Phase I | NCT00976729 | |
| Carlumab (anti-CCL2 antibodies Centocor) | CCL2 | Prostate cancer | Phase II | NCT00992186 | |
| CNTO 888 (Carlumab) | CCR2 | Prostate cancer | Phase II | NCT00992186 | |
| PF-04136309 | CCR2 | Pancreatic cancer | Phase I/II | NCT02732938 | |
| TAMs reprogramming | R848 | TLR7/8 | Colorectal cancer | Phase II | NCT00960752 |
| lefitolimod | TLR9 | Small-cell lung cancer | Phase I | NCT02668770 | |
| RP6530 | PI3Kδ/γ | Hodgkin lymphoma | Phase I/II | NCT03770000 | |
| Cell-based therapies |
CAR-M | HER2 | Solid cancers | Phase I | NCT04660929 |
| TEMFERON | Tie-2 | Glioblastoma multiforme | (Phase I/IIa) | NCT03866109 |
2.1. TAM Depletion
The depletion of TAM by clodronate [152], Zoledronate [15], Trabectedin [153], and FAK inhibitors, such as Defactinib, GSK2256098, and CT-707 (Conteltinib), has been found to inhibit tumor growth in solid cancers [154,155]. Clodronate encapsulated in liposomes effectively depletes macrophages in murine F9 teratocarcinoma and in human A673 rhabdomyosarcoma mouse tumor models and results in the significant inhibition of tumor growth [183]. Moreover, anti-tumor effects of bisphosphonates through TAM depletion have also been demonstrated in a 4T1 mouse breast cancer model [184]. In addition, the targeted TAM depletion or selective depletion of M2 TAMs can be achieved using innovative delivery methods. For example, zoledronate-loaded RBCs used in an innovative TAM-targeted delivery system showed promising results in mouse mammary carcinoma models [185]. Further, lipid-coated calcium zoledronate nanoparticles have shown selective targeting of M2-like TAMs and reduced tumor growth by the inhibition of immunosuppressive effects in a mouse model [15,186]. In another interesting strategy, the selective depletion of M2 TAM was achieved by using bi- and tri-valent T-cell engagers. Activation of endogenous T-cells by CD206- and FRβ-targeting BiTEs/TriTEs exerted preferential killing of M2- over M1-polarized macrophages [187]. Furthermore, TAM depletion by using CAR-T has recently been demonstrated in an orthotopic lung cancer mouse model. Chimeric antigen receptor (CAR) T-cells targeting the macrophage marker F4/80 (F4.CAR-T) effectively killed macrophages in vitro and in vivo without exerting toxicity. F4.CAR-T-cells infiltrated tumor lesions and delayed tumor growth comparably with a PD-1 blockade and significantly extended mouse survival [188]. However, despite these preclinical success stories, the durability of the response to TAM depletion remains below expectations. In addition, TAM depletion has been found to be less effective at preventing cancer growth as compared to the TAM-targeting approaches, such as blocking TAM, recruitment, or reprogramming.
2.2. Inhibition of Macrophage Recruitment
Macrophage accumulation in tumors is thought to be a result of the continuous inflow of monocytes from the circulation in response to tumor-derived factors. Colony-stimulating factor-1 (CSF-1) and C−C chemokine ligands, such as CCL2, are crucial tumor-derived factors that mediate the crosstalk between monocytes and tumor cells and foster continuous recruitment and differentiation of monocytes in the TME [189]. Therefore, blocking these factors would be an effective way to prevent the recruitment of TAMs into tumors. Several drugs targeting CSF-1/CSF-1R are currently being investigated in clinical trials (Table 1). PLX3397 (pexidartinib), a CSF-1R inhibitor, has recently been shown to improve the clinical symptoms of patients having Tenosynovial giant cell tumors (TGCT) in a randomized phase III trial [190]. In addition, a phase Ib study has shown a significant reduction in M2-like TAMs at tumor sites in patients with advanced solid tumors when treated with a combination of PLX3397 with paclitaxel [15,191]. In another Phase I/IIa trial, PLX3397 was evaluated in combination with pembrolizumab (PD-1 antibody) for the treatment of advanced melanoma and other solid tumors; however, the study was terminated early due to insufficient evidence of clinical efficacy (NCT02452424). Despite the therapeutic implications of CSF1R inhibitors, clinical trial outcomes based on CSF1R-blocking strategies have proven difficult to improve patient outcomes. Nevertheless, several small molecule and antibody-based CSF-1 inhibitors are also being tested in combination with other therapeutic modalities such as chemotherapy, radiotherapy, and immune checkpoint inhibitors (Table 2).
Table 2. Selected clinical trials of the TAM-targeting agents in combination with other therapeutic interventions.
| CSF-1R Inhibitors + Checkpoint Immunotherapy | ||||
|---|---|---|---|---|
| Drug Name | Combination Drugs | Cancer Types | Phasess | Clin. Trials |
| PLX3397 (Pexidarnitib) | Pembrolizumab | Solid tumors | Phase I/II | NCT02452424 |
| Durvalumab | Advanced tumors | Phase I | NCT02777710 | |
| LY3022855 mAb (IMC-CS4) | Pembrolizumab | Pancreatic cancer | Phase I | NCT03153410 |
| Durvalumab | ||||
| Tremelimumab | Advanced solid tumors | Phase I | NCT02718911 | |
| RO5509554/RG7155 (Emactuzumab) | Atezolizumab | Solid tumors | Phase I | NCT02323191 |
| CSF-1R Inhibitors + Chemotherapy | ||||
| PLX3397 (Pexidarnitib) | Paclitaxel | Advanced solid tumors | Phase I/II | NCT01525602 |
| Standard Chemotherapy | NCT01042379 | |||
| CSF-1R Inhibitors + Targeted Therapy | ||||
| PLX3397 (Pexidarnitib) | Sirolimus (Rapamycin) | Sarcoma | NCT02584647 | |
| CSF-1R Inhibitors + Radiotherapy | ||||
| PLX3397 (Pexidarnitib) | RT + ADT | Prostate cancer | Phase I | NCT02472275 |
| RT + Temozolomide | Glioblastoma | Phase I/II | NCT01790503 | |
| CCR2/CCR5 Inhibitors + Checkpoint Immunotherapy | ||||
| BMS-813160 (CCR2/CCR5 antagonist) | Nivolumab/Nabpaclitaxel | Advanced solid tumors | Phase I/II | NCT03184870 |
| Nivolumab/iplimumab | Phase II | NCT0299611 | ||
| Nivolumab | Hepatocellular carcinoma | Phase II | NCT04123379 | |
| CCR5 antagonist | Pembrolizumab | CRC | Phase I | NCT03274804 |
| Nivolumab plus Ipilimumab |
Pancreatic cancer, CRC | Phase I | NCT04721301 | |
| CCR2 Inhibitors + Chemotherapy | ||||
| CNTO 888 (Carlumab) | Gemcitabine/paclitaxel | Advanced solid tumors | Phase II | NCT01204996 |
| Carboplatin/doxorubicin | ||||
| PF-04136309 | FOLFIRINOX | Advanced solid tumors | Phase I/II | NCT01413022 |
| Anti-CD47/SIRPα antibodies+ Other immunotherapies | ||||
| Hu5F9-G4 | Pembrolizumab | Solid tumors | Phase I/II | NCT03869190 |
| Multiple immunotherapy | Urothelial and bladder cancer | Phase I/II | NCT03869190 | |
| BI 754,091 (OSE Immunotherapeutics) | BI 754,091 (anti-PD1) | Solid tumors | Phase I | NCT03990233 |
Likewise, targeting the CCL2-CCR2 axis also reduces the number of M2-like TAMs at primary and metastatic sites, increases CD8 T-cells, and inhibits tumor growth and invasion [192]. Multiple preclinical murine models have demonstrated the potent efficacy of CCR2 inhibitors and anti-CCL2 antibodies in reducing tumor growth and metastasis [193]. The CCL2 inhibitor mNOX-E36 inhibits the recruitment of M2-like TAMs and improves antiangiogenic treatment for glioblastoma in rats [194]. CCX872, another CCL2 antagonist, efficiently reduces tumor-associated MDSCs, which are converted into TAMs in the TME, thereby improving survival in animal models of glioblastoma [195]. A natural CCR2 antagonist (from Abies georgei and named 747) showed anti-tumor activity in mouse models of HCC. This was demonstrated by a reduction in the TAM level and concomitant expansion of CD8 T-cells in the TME [196]. In addition, the concurrent administration of anti-CCL2 antibodies was found to improve the efficacy of chemotherapy and checkpoint inhibitors [197] (Table 2). A phase Ib trial using PF-04136309, a CCR2 inhibitor, in combination with chemotherapy FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, oxaliplatin) in patients with borderline resectable and locally advanced pancreatic adenocarcinoma was found safe and tolerable, however, with a limited response [198]. Moreover, combining the CCR2 antagonist (RS504393) with anti-PD-1 resulted in an enhanced tumor response compared to anti-PD-1 monotherapy in multiple murine tumor models. This combination enhanced anti-tumor responses by increasing CD8+ T-cell recruitment and activation and decreasing CD4+ regulatory T-cells, simultaneously [199]. Moreover, in a preclinical study, a small molecule inhibitor of ASK1(MAP3K5), GS444217, was shown to inhibit ovarian cancer tumor growth by inhibiting the macrophage infiltration in peritoneal ascites by down modulating the activation of endothelial cells [182].
2.3. Inhibition of M2 Polarization and Reprogramming
As discussed above, monocytes recruited at the tumor site polarize into M2-like TAMs that promote tumor growth and progression. The reprogramming of M2-like TAMs to convert into M1-like subtypes is another therapeutic approach that is being tested. Puerarin, a MEK/ERK ½ inactivator, has been shown to inhibit M2 polarization in NSCLC [170]. HDAC inhibitors, such as Trichostatin-A [171] and TMP195 [172], promote the M1 polarization of TAMs. Recently, a pharmacological screening of small molecules identified that Doxycycline inhibits the polarization of macrophages towards the M2 phenotype, which in turn limits tumor growth by inhibiting neovascularization [173]. Similarly, STAT-3 inhibitors alone and in combination with the ERK inhibitor reduces TAM polarization [174]. In addition, bisphosphonate zoledronate can also repolarize the TAMs by targeting the mevalonate pathway and inhibiting the development of a mammary tumor [15]. Besides the inhibition of macrophage polarization towards an M2-type, the re-educating or reprogramming of M2 TAMs to behave like M1-type macrophages can also be an effective TAM-targeting strategy that has been focused on in recent years. RP-182, a synthetic peptide, reprograms M2 macrophages by activating the mannose receptor CD206 expressed on the M2 macrophage and turning them into M1-like phenotypes, thereby limiting tumor progression [175]. Duvelisib (IPI-145), an oral inhibitor of the PI3Kδ and PI3Kδγ isoforms, can induce the M2- to M1-like reprogramming of macrophages [176,177]. CD40 agonistic mAbs can induce TAMs to secrete high levels of matrix metalloproteinase 13 (MMP13), an enzyme that supports tumor control by degrading fibrotic tissue [178]. Multiple agonists of TLR7,8,9 have been shown to skew anti-tumor phenotypes in TAMs [179]. Clinical trials are also underway for TLR agonists to treat solid tumors (Table 1). TLR9 agonists, such as lefitolimod, effectively modulate the TME and induce anti-tumor responses by promoting the infiltration of CD8 T-cells and reprogramming TAMs in the TME. Ibrutinib, a Btk inhibitor has been shown to reprogram TAMs in pancreatic cancer [180]. In addition, the nanoparticles-based delivery of TLR agonists, bisphosphonates, DNA, mRNA, and miRNA repolarizes TAMs more effectively. A comprehensive review has recently been published on this topic [181].
2.4. Targeting Programmed Cell Removal (PrCR) and Anti-Phagocytic Checkpoints
Macrophages remove damaged, dysfunctional, aging, or harmful cells by phagocytosis called PrCR. The PrCR process involves recognizing, engulfing, and digesting target cells intracellularly. Several pro-phagocytic signals called “Eat me signals” have been identified that facilitate the PrCR of cancer cells. However, by expressing “Don’t eat me” signals, cancer cells counterbalance this elimination mechanism and evade immune clearance. An “Eat me signal” calreticulin secreted or exposed on the cell surface of cancer cells allows macrophages to recognize and phagocytose them [89,200]. However, this effect is antagonized by CD47, a “don’t eat me” signal often overexpressed by cancer cells. Through the interaction with its receptor, SIRPα, CD47 activates anti-inflammatory signaling in macrophages, causing them to cease phagocytosis. Further, PrCR is also induced by several chemotherapeutic agents, including docetaxel, doxorubicin, carboplatin, and cisplatin. This phenomenon is called immunological cell death. The dying tumor cells release tumor antigens and adjuvant molecules (ATP, HMGB1, and ecto-calreticulin) that trigger the involvement of macrophages in the immune response [164,165,166]. Docetaxel is a chemotherapeutic drug known for its antimitotic activity; however, recent studies have highlighted its immunomodulatory role. Docetaxel induces proinflammatory chemokine (C–C motif) ligand 3 (CCL3), which induces proinflammatory macrophage differentiation and phagocytosis [201]. In addition to this, docetaxel also induces calreticulin translocation to the plasma membrane of the dying cells and triggers phagocytosis of the cancer cells by TAMs. In a recent study, docetaxel and its combination with carboplatin or cisplatin significantly increased ATP levels, ecto-calreticulin expression, and HMGB1 expression in NSCLC cell lines, leading to the phagocytosis of treated cells and maturation of DCs [202]. Moreover, at the molecular level, TLR pathways and the subsequent activation of Bruton’s tyrosine kinase (Btk) signaling induces PrCR in macrophages by regulating the phosphorylation and surface trafficking of calreticulin [203]. Various therapeutic approaches to harness the ability of programmed cell removal by macrophages are in various stages of clinical development. A large number of inhibitors including monoclonal antibodies and soluble peptides directly targeting CD47 and SIRPα are in clinical trials. (Table 1). In addition, several other anti-phagocytic surface proteins (immune checkpoint) such as PDL1, LILRB1, B2M, and CD24 have also been found to inhibit the phagocytosis of cancer cells both in the in vitro co-culture of cancer cells with macrophages as well as in vivo. The inhibition of this immune checkpoint using monoclonal antibodies has shown promising results in preclinical studies. Some inhibitors such as (Anti-LILRB2) are also in clinical trials (Table 1).
2.5. Macrophages-Mediated Cytokine Delivery
A recent study by De Palma et al., used macrophages to deliver IFNγ to tumor sites. They transferred the Ifna1 gene into hematopoietic progenitors under the promoter of the Tie2 gene. Due to the high migratory and tumor-homing abilities of Tie2-expressing monocytes, the cell-specific expression of IFNγ in Tie2+ monocytes allowed the targeted release of IFNγ at the tumor sites [168]. The TME delivery of IFNγ inhibited tumor growth and angiogenesis by triggering the immune response. TEMFERON, genetically modified Tie2-expressing monocytes (TEMs), targeting interferon α-2 (IFNα-2) expression in GBM TME is being tested in a clinical trial (Table 1). Similarly, in another approach, IFNα containing soft particles called backpacks coated on the macrophage surface significantly reduced tumor growth and metastatic burden when injected intra-tumorally [169].
2.6. Macrophages-Based Adoptive Cell Transfusion
T lymphocyte-based adoptive cell therapies such as CAR-T-cells or TCR-engineered T-cells have shown remarkable anti-tumor responses in different advanced hematological malignancies, as evidenced by six FDA approvals in the last 10 years [204]. These therapeutic modalities are, however, not yet available in clinical trials for solid cancers. The low response of adoptive cell therapies in solid tumors is because of the limited penetration of adoptively transferred cells into the tumor sites. The barrier to this immune infiltration is usually immunosuppression and fibrotic tissue remodeling. As discussed above, TAMs are the major cause of tumor immune suppression and fibrosis; therefore, their depletion and inhibition of recruitment is being extensively investigated. However, unlike lymphocyte-based cellular therapeutic agents, macrophages have superior trafficking and homing capabilities in solid tumors which makes them a powerful therapeutic tool. Recently, Michael Klichinsky and colleagues pioneered in developing a CAR-M platform to treat HER2 positive solid tumors. The expression of CAR19ζ and HER2 ζ CARs in THP-1 cells specifically killed CD19+ K562 and HER2+ SKOV3, respectively, in an in vitro co-culture through enhanced phagocytosis. Further, a significantly reduced tumor growth and increases overall survival were demonstrated in two mice xenograft models and a humanized mouse model [151]. Currently, Carisma Therapeutics Inc. is conducting the first ever CAR-M clinical trial in humans for HER2 positive solid tumors, and it is presently in phase I (NCT04660929, recruiting).
3. Challenges in Therapeutic Targeting of TAMs
Macrophages are one of the most plastic cell types of the immune system. The local microenvironment in which they reside determines their polymorphism and functional heterogeneity. Macrophages constitute the most abundant population of immune origin in solid tumors. In a variety of human cancers, TAMs seem to be associated with a poor prognosis, although it varies by cancer type or context. The role played by TAMs in tumor progression makes them an attractive target for anti-tumor treatments [4,12,17,131]. There have been several therapeutic strategies developed that directly target TAMs or their functional mediators. These strategies include the depletion of TAMs, the blocking of monocyte recruitment, the reprogramming of TAMs into proinflammatory M1 macrophages, and the neutralization of their products [17,39,205]. Several antagonists that target TAMs have already been tested in various clinical trials, even though most TAM-targeting strategies are still in the preclinical stages [8,15,89,92,186,206,207,208,209,210]. There are still a lot of obstacles to overcome and many issues to be resolved before targeting TAMs becomes a reality. Finding the real prognostic value of TAMs in various cancers is a big challenge. There are disagreements among different studies regarding the prognostic value of TAMs in solid tumors. This is because different studies find different prognostic values of TAMs, not only depending on the cancer type, but also for the same type of cancer. For example, in the case of a highly heterogenous cancer such as gastric cancer, bladder cancer, and pancreatic cancer, both positive and negative prognostic values of TAMs have been reported [9,10,23,24,31,209]. These studies were limited by the poor understating of TAM heterogeneity and the use of M2 markers as the sole predictor. Hence, a further sub-typing of TAMs beyond an M1/M2 dichotomy can present a clearer picture of TAM density-based disease prognosis. Further, identifying the appropriate stage for TAM-targeted interventions to achieve the maximum therapeutic response can be achieved by better understanding the roles of TAMs in cancer progression from early-stage initiation to later metastatic stages. For example, a transition to M2 macrophages occurs in the advanced stages of cancer, whereas M1 macrophages dominate in the early stages [199]. Accordingly, TAM repolarization or M2 TAM depletion may be more effective during the advanced stages, whereas TAM activation may be more effective during the early stages when M1 macrophages dominate.
This entry is adapted from the peer-reviewed paper 10.3390/vaccines11010055
This entry is offline, you can click here to edit this entry!