There is a growing appreciation that the cells of the tumor microenvironment interact via variations in their dynamic regulation of mitochondrial function. The melatonergic pathway is a core aspect of mitochondrial function, with tumors 'domineering' the homeostasis established in the tumor microenvironment by regulating the core functioning of other cells. This is predominantly achieved by regulating the levels of melatonin and its immediate precursor, N-acetylserotonin (NAS). Applying melatonin to any tumor drives tumor death via apoptosis, whilst NAS can activate the trophic receptor, TrkB, to increase the survival and proliferation of tumors. Given such contrasting effects of melatonin and NAS, it is crucial for tumors to regulate the NAS/melatonin ratio within the tumor microenvironment. Two immune cells can readily kill cancer cells, namely natural killer (NK) cells and CD8+ t cells. The tumor inactivates these cells by releasing kynurenine, which activates the aryl hydrocarbon receptor (AhR) on these immune cells, leading to their inactivation ('exhaustion'), with AhR activation also increasing the NAS/melatonin ratio. NAS release enhances the survival and proliferation of cancer stem-like cells, thereby driving tumor maintenance and spread (metastasis). The cells that can readily kill cancer cells are therefore turned into providers of support for tumor survival and spread. This is achieved via the tumor's regulation of the mitochondrial melatonergic pathway. Similar tumor interactions with the mitochondrial melatonergic pathway allows the tumor to regulate other cells in the tumor microenvironment, as well as influencing how these cells interact with each other. This is predominantly achieved by the altered metabolism and mitochondrial function in the tumor shaping the mitochondrial function of other cells in the tumor microenvironment. As mitochondria evolved over evolution from ancient bacteria, these interactions across cell types in the tumor microenvironment may be viewed as evolutionary modified bacteria dynamically interacting with each other within a quest to achieve 'dominance' via the regulation of mitochondrial melatonergic pathway. This is a novel conceptualization of the tumor microenvironment that emphasizes core metabolic processes, and their regulation by the melatonergic pathway. As a frame of reference this allows the incorporation of previously disparate pieces of data on the tumor microenvironment and also provides a clearer pathway to new research, coupled to treatment implications.
- tumor microenvironment
- mitochondria
- melatonin
1. Introduction
2. Tumor Microenvironment
2.1. Tumour Microenvironment Cells
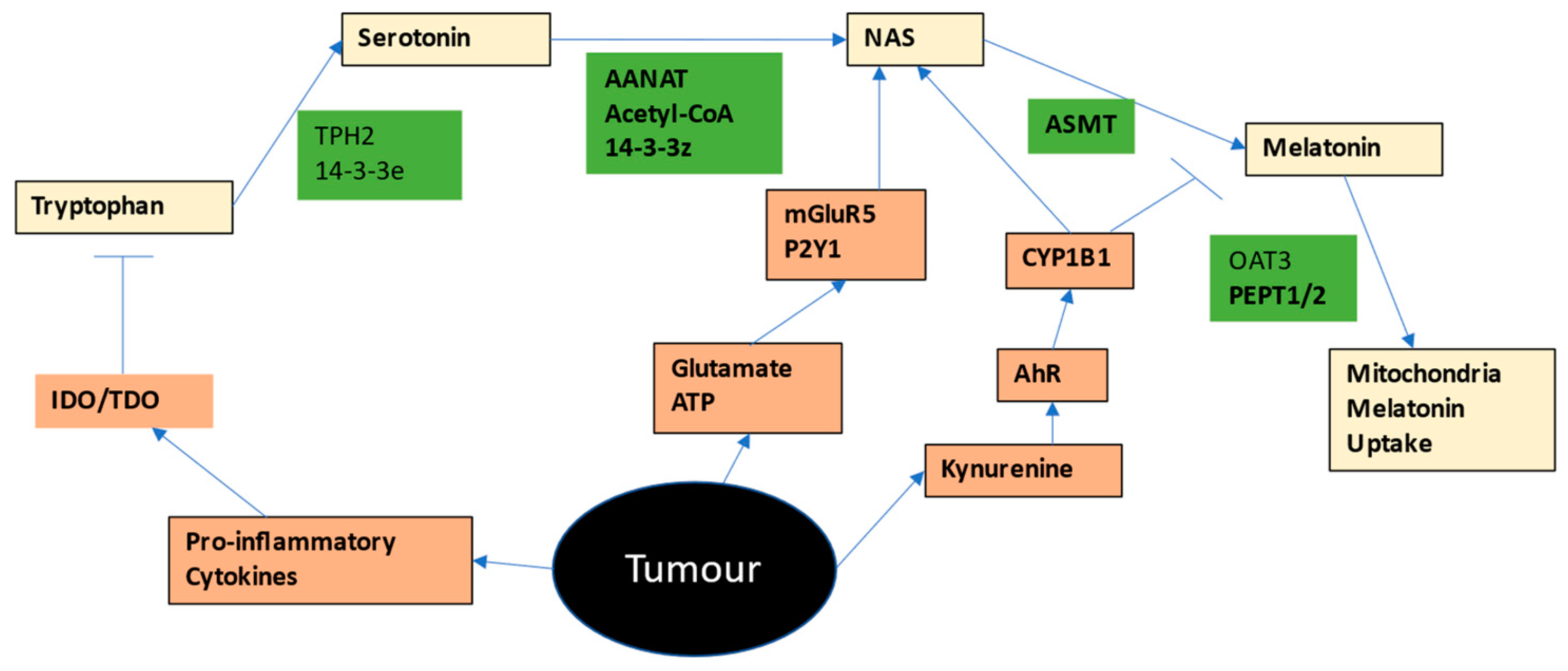
2.2. Tryptophan-Melatonin Pathway
2.3. Mitochondrial Metabolism in Tumour Microenvironment
2.3.1. Natural Killer Cells
2.3.2. Cytotoxic CD8+ t Cells
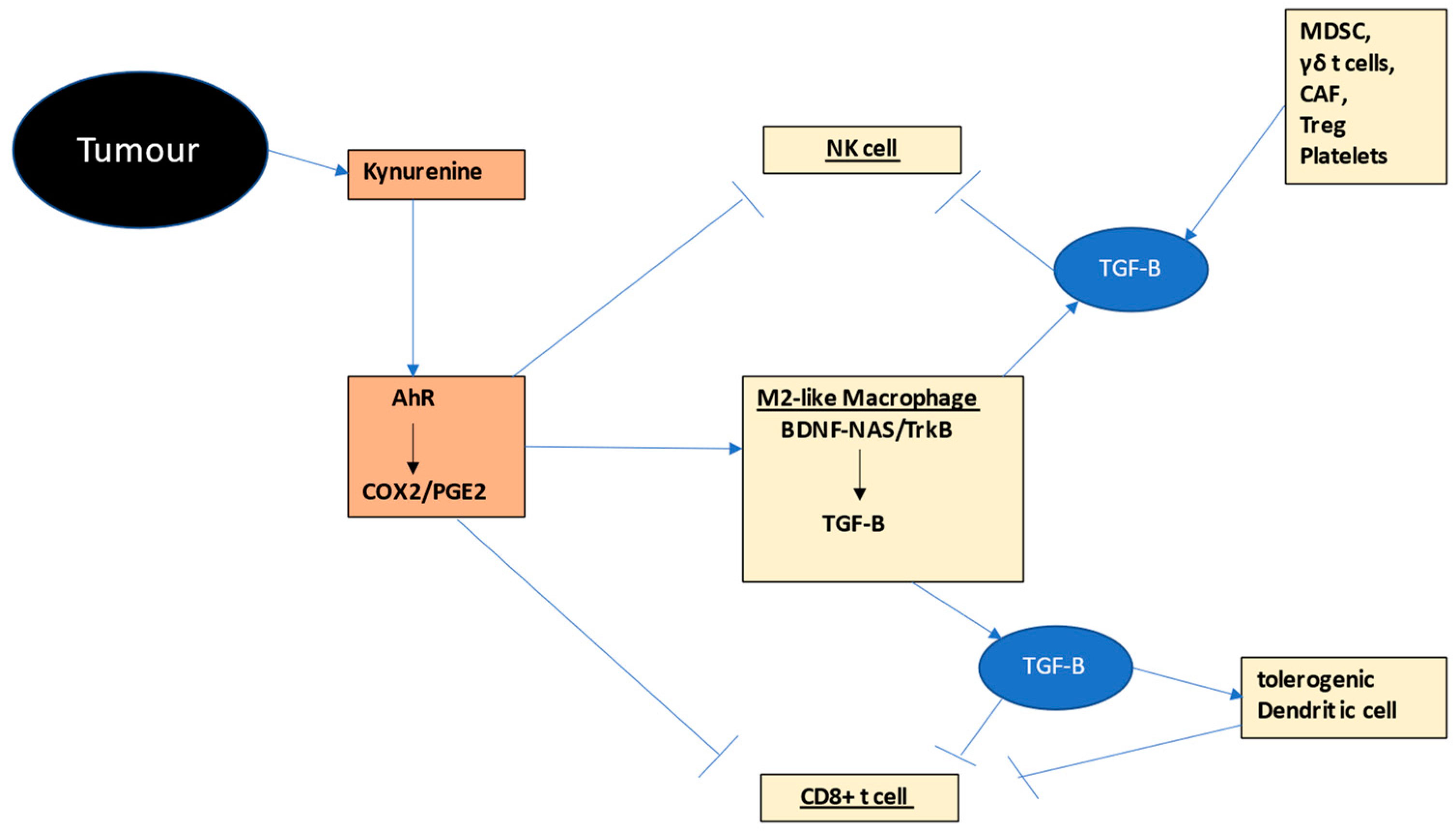
2.3.3. Macrophages
2.4. Neutrophils
2.5. Platelets
2.6. Other Tumour Microenvironment Cells
2.6.1. Myeloid-Derived Suppressor Cells (MDSCs)
2.6.2. Regulatory t Cells (Treg)
2.6.3. γδ t Cells
2.6.4. Dendritic Cells
2.6.5. Mesenchymal Stem Cells
2.7. Melatonergic Pathway and Immune Cells
2.8. MicroRNAs and Tumour Microenvironment
2.9. O-Linked-N-Acetylglucosaminylation (O-GlcNAcylation) and Tumour Microenvironment
This entry is adapted from the peer-reviewed paper 10.3390/ijms24010311
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530.
- Kobayashi, H. Recent advances in understanding the metabolic plasticity of ovarian cancer: A systematic review. Heliyon 2022, 8, e11487.
- Anderson, G. Tumour Microenvironment: Roles of the Aryl Hydrocarbon Receptor, O-GlcNAcylation, Acetyl-CoA and Melatonergic Pathway in Regulating Dynamic Metabolic Interactions across Cell Types—Tumour Microenvironment and Metabolism. Int. J. Mol. Sci. 2020, 22, 141.
- Muxel, S.M.; Pires-Lapa, M.A.; Monteiro, A.W.A.; Cecon, E.; Tamura, E.K.; Floeter-Winter, L.M.; Markus, R.P. NF-κB Drives the Synthesis of Melatonin in RAW 264.7 Macrophages by Inducing the Transcription of the Arylalkylamine-N-Acetyltransferase (AA-NAT) Gene. PLoS ONE 2012, 7, e52010.
- Markus, R.P.; Fernandes, P.A.; Kinker, G.S.; Cruz-Machado, S.D.S.; Marçola, M. Immune-pineal axis—Acute inflammatory responses coordinate melatonin synthesis by pinealocytes and phagocytes. Br. J. Pharmacol. 2017, 175, 3239–3250.
- Wang, S.; Sun, J.; Dastgheyb, R.M.; Li, Z. Tumor-derived extracellular vesicles modulate innate immune responses to affect tumor progression. Front. Immunol. 2022, 13, 1045624.
- Xavier, C.B.; Lopes, C.D.H.; Awni, B.M.; Campos, E.F.; Alves, J.P.B.; Camargo, A.A.; Guardia, G.D.A.; Galante, P.A.F.; Jardim, D.L. Interplay between Tumor Mutational Burden and Mutational Profile and Its Effect on Overall Survival: A Pilot Study of Metastatic Patients Treated with Immune Checkpoint Inhibitors. Cancers 2022, 14, 5433.
- Mesnage, R.; Antoniou, M.N. Computational modelling provides insight into the effects of glyphosate on the shikimate pathway in the human gut microbiome. Curr. Res. Toxicol. 2020, 1, 25–33.
- Huo, X.; Wang, C.; Yu, Z.; Peng, Y.; Wang, S.; Feng, S.; Zhang, S.; Tian, X.; Sun, C.; Liu, K.; et al. Human transporters, PEPT1/2, facilitate melatonin transportation into mitochondria of cancer cells: An implication of the therapeutic potential. J. Pineal Res. 2017, 62, e12390.
- Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500.
- Pagan, C.; Goubran-Botros, H.; Delorme, R.; Benabou, M.; Lemière, N.; Murray, K.; Amsellem, F.; Callebert, J.; Chaste, P.; Jamain, S.; et al. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci. Rep. 2017, 7, 2096.
- Qiu, J.; Zhang, J.; Zhou, Y.; Li, X.; Li, H.; Liu, J.; Gou, K.; Zhao, J.; Cui, S. MicroRNA-7 inhibits melatonin synthesis by acting as a linking molecule between leptin and norepinephrine signaling pathways in pig pineal gland. J. Pineal Res. 2019, 66, e12552.
- Su, X.; Wang, W.; Ma, S.; Ning, H.; Chen, J. Regulation effect of miR-7 on intervening colorectal cancer rats with HP infection through Akt/GSK-3β/β-catenin pathway. Cell. Mol. Biol. 2022, 68, 135–139.
- Wu, K.; Liu, F.; Zhang, T.; Zhou, Z.; Yu, S.; Quan, Y.; Zhu, S. miR-375 suppresses the growth and metastasis of esophageal squamous cell carcinoma by targeting PRDX1. Cell Mol. Biol. 2022, 13, 2154–2168.
- Du, Y.; Miao, Z.; Qiu, L.; Lv, Y.; Wang, K.; Guo, L. Clinical Potential of miR-451 and miR-506 as a Prognostic Biomarker in Patients with Breast Cancer. J. Healthc. Eng. 2022, 2022, 9578788.
- Le, H.T.T.; Murugesan, A.; Candeias, N.R.; Yli-Harja, O.; Kandhavelu, M. Functional characterization of HIC, a P2Y1 agonist, as a p53 stabilizer for prostate cancer cell death induction. Future Med. Chem. 2021, 13, 1845–1864.
- Kou, W.; Huang, H.; Dai, S.; Tan, X.; Chen, Q.; Huang, R.; Zou, H. mGluR5 promotes the progression of multiple myeloma in vitro via Ras–MAPK signaling pathway. Adv. Clin. Exp. Med. 2022, 31, 881–888.
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.
- Kanno, Y.; Saito, N.; Yamashita, N.; Ota, K.; Shizu, R.; Hosaka, T.; Nemoto, K.; Yoshinari, K. Possible Involvement of the Upregulation of ΔNp63 Expression Mediated by HER2-Activated Aryl Hydrocarbon Receptor in Mammosphere Maintenance. Int. J. Mol. Sci. 2022, 23, 12095.
- Li, M.; Hao, B.; Zhang, M.; Reiter, R.J.; Lin, S.; Zheng, T.; Chen, X.; Ren, Y.; Yue, L.; Abay, B.; et al. Melatonin enhances radiofrequency-induced NK antitumor immunity, causing cancer metabolism reprogramming and inhibition of multiple pulmonary tumor development. Signal Transduct. Target. Ther. 2021, 6, 330.
- Zhang, Z.; Du, J.; Xu, Q.; Xing, C.; Li, Y.; Zhou, S.; Zhao, Z.; Mu, Y.; Zhao, Z.; Cao, S.; et al. Adiponectin Suppresses Metastasis of Nasopharyngeal Carcinoma through Blocking the Activation of NF-κB and STAT3 Signaling. Int. J. Mol. Sci. 2022, 23, 12729.
- Fu, X.; Ji, F.; He, Q.; Qiu, X. A Systematic Pan-Cancer Analysis of YY1 Aberrations and their Relationship with Clinical Outcome, Tumor Microenvironment, and Therapeutic Targets. J. Immunol. Res. 2022, 2022, 5826741.
- Bernard, M.; Voisin, P. Photoreceptor-specific expression, light-dependent localization, and transcriptional targets of the zinc-finger protein Yin Yang 1 in the chicken retina. J. Neurochem. 2008, 105, 595–604.
- Anderson, G.; Reiter, R.J. Glioblastoma: Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Mediating Effects of miR-451 and Aryl Hydrocarbon Receptor and in Coordinating Wider Biochemical Changes. Int. J. Tryptophan Res. 2019, 12, 1178646919855942.
- Jang, S.-W.; Liu, X.; Pradoldej, S.; Tosini, G.; Chang, Q.; Iuvone, P.M.; Ye, K. N -acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA 2010, 107, 3876–3881.
- Malekan, M.; Nezamabadi, S.S.; Samami, E.; Mohebalizadeh, M.; Saghazadeh, A.; Rezaei, N. BDNF and its signaling in cancer. J. Cancer Res. Clin. Oncol. 2022, 1–16.
- Moriwaki, K.; Wada, M.; Kuwabara, H.; Ayani, Y.; Terada, T.; Higashino, M.; Kawata, R.; Asahi, M. BDNF/TRKB axis provokes EMT progression to induce cell aggressiveness via crosstalk with cancer-associated fibroblasts in human parotid gland cancer. Sci. Rep. 2022, 12, 17553.
- Samanta, S. Melatonin: A Potential Antineoplastic Agent in Breast Cancer. J. Environ. Pathol. Toxicol. Oncol. 2022, 41, 55–84.
- Cucielo, M.S.; Cesário, R.C.; Silveira, H.S.; Gaiotte, L.B.; dos Santos, S.A.A.; Zuccari, D.A.P.D.C.; Seiva, F.R.F.; Reiter, R.J.; Chuffa, L.G.D.A. Melatonin Reverses the Warburg-Type Metabolism and Reduces Mitochondrial Membrane Potential of Ovarian Cancer Cells Independent of MT1 Receptor Activation. Molecules 2022, 27, 4350.
- Luo, X.; Chen, Y.; Tang, H.; Wang, H.; Jiang, E.; Shao, Z.; Liu, K.; Zhou, X.; Shang, Z. Melatonin inhibits EMT and PD-L1 expression through the ERK1/2/FOSL1 pathway and regulates anti-tumor immunity in HNSCC. Cancer Sci. 2022, 113, 2232–2245.
- Tan, D.-X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2012, 54, 127–138.
- Anderson, G.; Carbone, A.; Mazzoccoli, G. Tryptophan Metabolites and Aryl Hydrocarbon Receptor in Severe Acute Respiratory Syndrome, Coronavirus-2 (SARS-CoV-2) Pathophysiology. Int. J. Mol. Sci. 2021, 22, 1597.
- ToVinh, M.; Hörr, G.; Dobrikova, K.; Gotter, C.; Rommel, C.; Hoffmeister, C.; Raabe, J.; Kaiser, K.M.; Finnemann, C.; Bischoff, J.; et al. Mitochondrial Dysfunction Contributes to Impaired Cytokine Production of CD56bright Natural Killer Cells from Human Immunodeficiency Virus–Infected Individuals Under Effective Antiretroviral Therapy. J. Infect. Dis. 2022, 226, 901–906.
- Slattery, K.; Woods, E.; Zaiatz-Bittencourt, V.; Marks, S.; Chew, S.; Conroy, M.; Goggin, C.; MacEochagain, C.; Kennedy, J.; Lucas, S.; et al. TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J. Immunother. Cancer 2021, 9, e002044.
- Jiang, B.; Kang, X.; Zhao, G.; Lu, J.; Wang, Z. miR-138 Reduces the Dysfunction of T Follicular Helper Cells in Osteosarcoma via the PI3K/Akt/mTOR Pathway by Targeting PDK1. Comput. Math. Methods Med. 2021, 2021, 2895893.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rorsales-Corral, S.; de Almeida Chuffa, L.G. Melatonin inhibits Warburg-dependent cancer by redirecting glucose oxidation to the mitochondria: A mechanistic hypothesis. Cell. Mol. Life Sci. 2020, 77, 2527–2542.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: A proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–119.
- Bittencourt, V.Z.; Jones, F.; Tosetto, M.; A Doherty, G.; Ryan, E.J. Dysregulation of Metabolic Pathways in Circulating Natural Killer Cells Isolated from Inflammatory Bowel Disease Patients. J. Crohn’s Colitis 2021, 15, 1316–1325.
- Calvo, J.R.; González-Yanes, C.; Maldonado, M.D. The role of melatonin in the cells of the innate immunity: A review. J. Pineal Res. 2013, 55, 103–120.
- Perfilyeva, Y.; Ostapchuk, Y.O.; Abdolla, N.; Tleulieva, R.; Krasnoshtanov, V.C.; Belyaev, N.N. Exogenous Melatonin Up-Regulates Expression of CD62L by Lymphocytes in Aged Mice under Inflammatory and Non-Inflammatory Conditions. Immunol. Investig. 2019, 48, 632–643.
- Lansdorp, P.M. Telomeres, Telomerase and Cancer. Arch. Med. Res. 2022, 53, 741–746.
- Arjona, A.; Sarkar, D.K. Circadian Oscillations of Clock Genes, Cytolytic Factors, and Cytokines in Rat NK Cells. J. Immunol. 2005, 174, 7618–7624.
- Mortezaee, K.; Potes, Y.; Mirtavoos-Mahyari, H.; Motevaseli, E.; Shabeeb, D.; Musa, A.E.; Najafi, M.; Farhood, B. Boosting immune system against cancer by melatonin: A mechanistic viewpoint. Life Sci. 2019, 238, 116960.
- Lisci, M.; Barton, P.R.; Randzavola, L.O.; Ma, C.Y.; Marchingo, J.M.; Cantrell, D.A.; Paupe, V.; Prudent, J.; Stinchcombe, J.C.; Griffiths, G.M. Mitochondrial translation is required for sustained killing by cytotoxic T cells. Science 2021, 374, eabe9977.
- Mougiakakos, D.; Kahlfuss, S. “Moonlighting” at the power plant: How mitochondria facilitate serial killing by CD8+ cytotoxic T cells. Signal Transduct. Target. Ther. 2021, 6, 436.
- Nobis, C.C.; Laramée, G.D.; Kervezee, L.; De Sousa, D.M.; Labrecque, N.; Cermakian, N. The circadian clock of CD8 T cells modulates their early response to vaccination and the rhythmicity of related signaling pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 20077–20086.
- Rahimi, S.B.; Mohebbi, A.; Vakilzadeh, G.; Biglari, P.; Jahromi, S.R.; Mohebi, S.R.; Shirian, S.; Gorji, A.; Ghaemi, A. Enhancement of therapeutic DNA vaccine potency by melatonin through inhibiting VEGF expression and induction of antitumor immunity mediated by CD8+ T cells. Arch. Virol. 2017, 163, 587–597.
- Huang, X.; Wang, L.; Guo, H.; Zhang, W. Macrophage membrane-coated nanovesicles for dual-targeted drug delivery to inhibit tumor and induce macrophage polarization. Bioact. Mater. 2022, 23, 69–79.
- Kawaguchi, Y.; Ohshio, Y.; Watanabe, A.; Shiratori, T.; Okamoto, K.; Ueda, K.; Kataoka, Y.; Suzuki, T.; Hanaoka, J. Depletion of tumor-associated macrophages inhibits the lung cancer growth and enhances the antitumor effect of cisplatin. Cancer Sci. 2022, in press.
- Xun, X.; Zhang, C.; Wang, S.; Hu, S.; Xiang, X.; Cheng, Q.; Li, Z.; Wang, Y.; Zhu, J. Cyclooxygenase-2 expressed hepatocellular carcinoma induces cytotoxic T lymphocytes exhaustion through M2 macrophage polarization. Am. J. Transl. Res. 2021, 13, 4360–4375.
- Bi, C.; Fu, Y.; Zhang, Z.; Li, B. Prostaglandin E2 confers protection against diabetic coronary atherosclerosis by stimulating M2 macrophage polarization via the activation of the CREB/BDNF/TrkB signaling pathway. FASEB J. 2020, 34, 7360–7371.
- Huang, Z.-B.; Hu, Z.; Lu, C.-X.; Luo, S.-D.; Chen, Y.; Zhou, Z.-P.; Hu, J.-J.; Zhang, F.-L.; Deng, F.; Liu, K.-X. Gut microbiota-derived indole 3-propionic acid partially activates aryl hydrocarbon receptor to promote macrophage phagocytosis and attenuate septic injury. Front. Cell. Infect. Microbiol. 2022, 12, 1015386.
- Park, S.-H.; Yoon, S.-J.; Choi, S.; Jung, J.; Park, J.-Y.; Park, Y.-H.; Seo, J.; Lee, J.; Lee, M.-S.; Lee, S.-J.; et al. Particulate matter promotes cancer metastasis through increased HBEGF expression in macrophages. Exp. Mol. Med. 2022, 54, 1901–1912.
- Selvam, P.; Cheng, C.-M.; Dahms, H.-U.; Ponnusamy, V.K.; Sun, Y.-Y. AhR Mediated Activation of Pro-Inflammatory Response of RAW 264. 7 Cells Modul. Epithel.-Mesenchymal Transit. 2022, 10, 642.
- Aria, H.; Rezaei, M.; Nazem, S.; Daraei, A.; Nikfar, G.; Mansoori, B.; Bahmanyar, M.; Tavassoli, A.; Vakil, M.K.; Mansoori, Y. Purinergic receptors are a key bottleneck in tumor metabolic reprogramming: The prime suspect in cancer therapeutic resistance. Front. Immunol. 2022, 13, 947885.
- Merz, J.; Nettesheim, A.; von Garlen, S.; Albrecht, P.; Saller, B.S.; Engelmann, J.; Hertle, L.; Schäfer, I.; Dimanski, D.; König, S.; et al. Pro- and anti-inflammatory macrophages express a sub-type specific purinergic receptor profile. Purinergic Signal. 2021, 17, 481–492.
- Xiong, T.; He, P.; Zhou, M.; Zhong, D.; Yang, T.; He, W.; Xu, Z.; Chen, Z.; Liu, Y.; Dai, S. Glutamate blunts cell-killing effects of neutrophils in tumor microenvironment. Cancer Sci. 2022, 113, 1955–1967.
- Shanshiashvili, L.; Tsitsilashvili, E.; Dabrundashvili, N.; Kalandadze, I.; Mikeladze, D. Metabotropic glutamate receptor 5 may be involved in macrophage plasticity. Biol. Res. 2017, 50, 4.
- Zhou, J.; Liu, H.; Jiang, S.; Wang, W. Role of tumor-associated neutrophils in lung cancer (Review). Oncol Lett. 2022, 25, 2.
- Bock, K.W. Aryl hydrocarbon receptor (AHR) functions: Balancing opposing processes including inflammatory reactions. Biochem. Pharmacol. 2020, 178, 114093.
- Liu, X.-Y.; Liu, Y.; Li, J.-F.; Yue, S.-J.; Shen, L.; Li, C.; Han, J.-Z.; Xu, J.-P.; Feng, D.-D.; Liu, H.-J.; et al. Activation of mGluRI in neutrophils promotes the adherence of neutrophils to endothelial cells. Sheng Li Xue Bao 2010, 62, 219–224.
- Collard, C.D.; Park, K.A.; Montalto, M.C.; Alapati, S.; Buras, J.A.; Stahl, G.L.; Colgan, S.P. Neutrophil-derived Glutamate Regulates Vascular Endothelial Barrier Function. J. Biol. Chem. 2002, 277, 14801–14811.
- Zhang, X.; Zhao, W.; Zhao, Y.; Zhao, Z.; Lv, Z.; Zhang, Z.; Ren, H.; Wang, Q.; Liu, M.; Qian, M.; et al. Inflammatory macrophages exacerbate neutrophil-driven joint damage through ADP/P2Y1 signaling in rheumatoid arthritis. Sci. China Life Sci. 2021, 65, 953–968.
- Cao, Z.; Zhao, M.; Sun, H.; Hu, L.; Chen, Y.; Fan, Z. Roles of mitochondria in neutrophils. Front. Immunol. 2022, 13, 934444.
- Peng, S.; Gao, J.; Stojkov, D.; Yousefi, S.; Simon, H.U. Established and emerging roles for mitochondria in neutrophils. Immunol. Rev. 2022.
- Sousa, W.O.; Fujimori, M.; Morais, T.C.; Santos, M.B.; Rodrigues, G.F.S.; Silva, K.P.G.; Torres, A.H.F.; Honorio-França, A.C.; França, E.L. Effects of Modified Melatonin Release on Human Colostrum Neutrophils to Induce Death in the MCF-7 Cell Line. Int. J. Cell Biol. 2022, 2022, 8069188.
- Naveenkumar, S.K.; Hemshekhar, M.; Jagadish, S.; Manikanta, K.; Ks, G.; Kemparaju, K.; Girish, K.S. Melatonin restores neutrophil functions and prevents apoptosis amid dysfunctional glutathione redox system. J. Pineal Res. 2020, 69, e12676.
- Sakakura, Y.; Sato, H.; Shiiya, A.; Tamba, M.; Sagara, J.-I.; Matsuda, M.; Okamura, N.; Makino, N.; Bannai, S. Expression and function of cystine/glutamate transporter in neutrophils. J. Leukoc. Biol. 2007, 81, 974–982.
- Xu, L.; Zhang, W.; Kwak, M.; Zhang, L.; Lee, P.C.W.; Jin, J.-O. Protective Effect of Melatonin Against Polymicrobial Sepsis Is Mediated by the Anti-bacterial Effect of Neutrophils. Front. Immunol. 2019, 10, 1371.
- Hou, Y.; Yang, D.; Xiang, R.; Wang, H.; Wang, X.; Zhang, H.; Wang, P.; Zhang, Z.; Che, X.; Liu, Y.; et al. N2 neutrophils may participate in spontaneous recovery after transient cerebral ischemia by inhibiting ischemic neuron injury in rats. Int. Immunopharmacol. 2019, 77, 105970.
- Zheng, W.; Fan, X.; Yang, Z.; Shangguan, Y.; Jin, T.; Liu, Y.; Huang, J.; Ye, X.; Zhou, Q.; Li, X. Strong inflammatory signatures in the neutrophils of PAMI syndrome. Front. Immunol. 2022, 13, 926087.
- Lin, J.; He, Y.; Wang, B.; Xun, Z.; Chen, S.; Zeng, Z.; Ou, Q. Blocking of YY1 reduce neutrophil infiltration by inhibiting IL-8 production via the PI3K-Akt-mTOR signaling pathway in rheumatoid arthritis. Clin. Exp. Immunol. 2018, 195, 226–236.
- Léger, J.L.; Soucy, M.N.; Veilleux, V.; Foulem, R.D.; A Robichaud, G.; E Surette, M.; Allain, E.P.; Boudreau, L.H. Functional platelet-derived mitochondria induce the release of human neutrophil microvesicles. EMBO Rep. 2022, 23, e54910.
- Strasenburg, W.; Jóźwicki, J.; Durślewicz, J.; Kuffel, B.; Kulczyk, M.P.; Kowalewski, A.; Grzanka, D.; Drewa, T.; Adamowicz, J. Tumor Cell-Induced Platelet Aggregation as an Emerging Therapeutic Target for Cancer Therapy. Front. Oncol. 2022, 12, 909767.
- Filippelli, A.; Del Gaudio, C.; Simonis, V.; Ciccone, V.; Spini, A.; Donnini, S. Scoping Review on Platelets and Tumor Angiogenesis: Do We Need More Evidence or Better Analysis? Int. J. Mol. Sci. 2022, 23, 13401.
- Pan, Y.; Wang, Y.; Wang, Y.; Xu, S.; Jiang, F.; Han, Y.; Hu, M.; Liu, Z. Platelet-derived microvesicles (PMVs) in cancer progression and clinical applications. Clin. Transl. Oncol. 2022, 1–9.
- Cho, M.S.; Lee, H.; Gonzalez-Delgado, R.; Li, D.; Sasano, T.; Carlos-Alcalde, W.; Ma, Q.; Liu, J.; Sood, A.K.; Afshar-Kharghan, V. Platelets Increase the Expression of PD-L1 in Ovarian Cancer. Cancers 2022, 14, 2498.
- Shu, Y.; Peng, J.; Feng, Z.; Hu, K.; Li, T.; Zhu, P.; Cheng, T.; Hao, L. Osteosarcoma subtypes based on platelet-related genes and tumor microenvironment characteristics. Front. Oncol. 2022, 12, 941724.
- Yuan, M.; Jia, Y.; Xing, Y.; Wang, Y.; Liu, Y.; Liu, X.; Liu, D. Screening and validation of platelet activation-related lncRNAs as potential biomarkers for prognosis and immunotherapy in gastric cancer patients. Front. Genet. 2022, 13, 965033.
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.-B.; Lee, S.K.; Bach, D.-H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers 2019, 11, 240.
- Amison, R.; Arnold, S.; O’Shaughnessy, B.; Cleary, S.; Ofoedu, J.; Idzko, M.; Page, C.; Pitchford, S. Lipopolysaccharide (LPS) induced pulmonary neutrophil recruitment and platelet activation is mediated via the P2Y1 and P2Y14 receptors in mice. Pulm. Pharmacol. Ther. 2017, 45, 62–68.
- Kim, D.; Shin, D.-Y.; Liu, J.; Jeong, N.-R.; Koh, Y.; Hong, J.; Huang, X.; Broxmeyer, H.E.; Yoon, S.-S. Expansion of Human Megakaryocyte-Lineage Progeny via Aryl Hydrocarbon Receptor Antagonism with CH223191. Stem Cell Rev. Rep. 2022, 18, 2982–2994.
- Pombo, M.; Lamé, M.W.; Walker, N.J.; Huynh, D.H.; Tablin, F. TCDD and omeprazole prime platelets through the aryl hydrocarbon receptor (AhR) non-genomic pathway. Toxicol. Lett. 2015, 235, 28–36.
- Schneider, M.A.; Heeb, L.; Beffinger, M.M.; Pantelyushin, S.; Linecker, M.; Roth, L.; Lehmann, K.; Ungethüm, U.; Kobold, S.; Graf, R.; et al. Attenuation of peripheral serotonin inhibits tumor growth and enhances immune checkpoint blockade therapy in murine tumor models. Sci. Transl. Med. 2021, 13, eabc8188.
- Sonkar, V.K.; Eustes, A.S.; Ahmed, A.; Jensen, M.; Solanki, M.V.; Swamy, J.; Kumar, R.; Fidler, T.P.; Houtman, J.C.; Allen, B.G.; et al. Endogenous SOD2 (Superoxide Dismutase) Regulates Platelet-Dependent Thrombin Generation and Thrombosis During Aging. Arter. Thromb. Vasc. Biol. 2022, 43, 79–91.
- Boilard, E.; Bellio, M. Platelet extracellular vesicles and the secretory interactome join forces in health and disease. Immunol. Rev. 2022, 312, 38–51.
- Xue, Y.; Zhang, L.; Zhang, L.; Sun, W.; Fang, Z.; Leng, Y.; Li, M.; Ren, X.; Zhang, R.; Zhang, Y.; et al. Danshensu prevents thrombosis by inhibiting platelet activation via SIRT1/ROS/mtDNA pathways without increasing bleeding risk. Phytomedicine 2022, 104, 154271.
- Kaczara, P.; Sitek, B.; Przyborowski, K.; Kurpinska, A.; Kus, K.; Stojak, M.; Chlopicki, S. Antiplatelet Effect of Carbon Monoxide Is Mediated by NAD + and ATP Depletion. Arter. Thromb. Vasc. Biol. 2020, 40, 2376–2390.
- Kulkarni, P.; Tiwari, A.; Singh, N.; Gautam, D.; Sonkar, V.K.; Agarwal, V.; Dash, D. Aerobic glycolysis fuels platelet activation: Small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica 2018, 104, 806–818.
- Yang, M.; Li, L.; Chen, S.; Li, S.; Wang, B.; Zhang, C.; Chen, Y.; Yang, L.; Xin, H.; Chen, C.; et al. Melatonin protects against apoptosis of megakaryocytic cells via its receptors and the AKT/mitochondrial/caspase pathway. Aging 2020, 12, 13633–13646.
- Van Faassen, M.; Peters, M.A.; Walenkamp, A.M.; de Vries, E.G.; Oosting, S.F.; Kema, I.P. Melatonin is not stored in platelets. Clin. Chim. Acta 2019, 498, 17–20.
- Boukhatem, I.; Fleury, S.; Welman, M.; Le Blanc, J.; Thys, C.; Freson, K.; Best, M.G.; Würdinger, T.; Allen, B.G.; Lordkipanidzé, M. The brain-derived neurotrophic factor prompts platelet aggregation and secretion. Blood Adv. 2021, 5, 3568–3580.
- Yoo, D.Y.; Nam, S.M.; Kim, W.; Lee, C.H.; Won, M.-H.; Hwang, I.K.; Yoon, Y.S. N-Acetylserotonin Increases Cell Proliferation and Differentiating Neuroblasts with Tertiary Dendrites through Upregulation of Brain-Derived Neurotrophic Factor in the Mouse Dentate Gyrus. J. Veter.-Med. Sci. 2011, 73, 1411–1416.
- Xie, W.; Xiang, L.; Song, Y.; Tian, X. The Downregulation of Truncated TrkB Receptors Modulated by MicroRNA-185 Activates Full-Length TrkB Signaling and Suppresses the Epileptiform Discharges in Cultured Hippocampal Neurons. Neurochem. Res. 2020, 45, 1647–1660.
- Anderson, G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut microbiome and muscle interactions via the Mitochondrial melatonergic pathway, with disruption by Glyphosate-based herbicides. Int. J. Mol. Sci. in press.
- Anderson, G.; Maes, M. Melatonin: A Natural Homeostatic Regulator–Interactions with Immune Inflammation and Trytophan Catabolite Pathways in the Modulation of Migraine and Endometriosis. J. Nat. Prod. Res. Updat. 2015, 1, 7–17.
- Beischlag, T.V.; Anderson, G.; Mazzoccoli, G. Glioma: Tryptophan Catabolite and Melatoninergic Pathways Link microRNA, 14-3- 3, Chromosome 4q35, Epigenetic Processes and other Glioma Biochemical Changes. Curr. Pharm. Des. 2016, 22, 1033–1048.
- Le Tong, L.; Jiménez-Cortegana, C.; Tay, A.H.; Wickström, S.; Galluzzi, L.; Lundqvist, A. NK cells and solid tumors: Therapeutic potential and persisting obstacles. Mol. Cancer 2022, 21, 206.
- Li, X.; Li, Y.; Yu, Q.; Qian, P.; Huang, H.; Lin, Y. Metabolic reprogramming of myeloid-derived suppressor cells: An innovative approach confronting challenges. J. Leukoc. Biol. 2021, 110, 257–270.
- Neamah, W.H.; Singh, N.P.; Alghetaa, H.; Abdulla, O.; Chatterjee, S.; Busbee, P.B.; Nagarkatti, M.; Nagarkatti, P. AhR Activation Leads to Massive Mobilization of Myeloid-Derived Suppressor Cells with Immunosuppressive Activity through Regulation of CXCR2 and MicroRNA miR-150-5p and miR-543-3p That Target Anti-Inflammatory Genes. J. Immunol. 2019, 203, 1830–1844.
- Wei, Y.; Na Peng, N.; Deng, C.; Zhao, F.; Tian, J.; Tang, Y.; Yu, S.; Chen, Y.; Xue, Y.; Xiao, F.; et al. Aryl hydrocarbon receptor activation drives polymorphonuclear myeloid-derived suppressor cell response and efficiently attenuates experimental Sjögren’s syndrome. Cell Mol. Immunol. 2022, 19, 1361–1372.
- Cao, W.; Lu, J.; Li, L.; Qiu, C.; Qin, X.; Wang, T.; Li, S.; Zhang, J.; Xu, J. Activation of the Aryl Hydrocarbon Receptor Ameliorates Acute Rejection of Rat Liver Transplantation by Regulating Treg Proliferation and PD-1 Expression. Transplantation 2022, 106, 2172–2181.
- Wu, H.; Herr, D.; MacIver, N.J.; Rathmell, J.C.; Gerriets, V.A. CD4 T cells differentially express cellular machinery for serotonin signaling, synthesis, and metabolism. Int. Immunopharmacol. 2020, 88, 106922.
- Wang, X.; Shen, H.; Zhangyuan, G.; Huang, R.; Zhang, W.; He, Q.; Jin, K.; Zhuo, H.; Zhang, Z.; Wang, J.; et al. 14-3-3ζ delivered by hepatocellular carcinoma-derived exosomes impaired anti-tumor function of tumor-infiltrating T lymphocytes. Cell Death Dis. 2018, 9, 159.
- Glebezdina, N.S.; Olina, A.A.; Nekrasova, I.V.; Kuklina, E.M. Molecular Mechanisms of Control of Differentiation of Regulatory T-Lymphocytes by Exogenous Melatonin. Dokl. Biochem. Biophys. 2019, 484, 13–16.
- Corsale, A.M.; Di Simone, M.; Presti, E.L.; Picone, C.; Dieli, F.; Meraviglia, S. Metabolic Changes in Tumor Microenvironment: How Could They Affect γδ T Cells Functions? Cells 2021, 10, 2896.
- Chen, D.; Guo, Y.; Jiang, J.; Wu, P.; Zhang, T.; Wei, Q.; Huang, J.; Wu, D. γδ T cell exhaustion: Opportunities for intervention. J. Leukoc. Biol. 2022, 112, 1669–1676.
- Dupraz, L.; Magniez, A.; Rolhion, N.; Richard, M.L.; Da Costa, G.; Touch, S.; Mayeur, C.; Planchais, J.; Agus, A.; Danne, C.; et al. Gut microbiota-derived short-chain fatty acids regulate IL-17 production by mouse and human intestinal γδ T cells. Cell Rep. 2021, 36, 109332.
- Kouketsu, A.; Haruka, S.; Kuroda, K.; Hitoshi, M.; Kensuke, Y.; Tsuyoshi, S.; Takahashi, T.; Hiroyuki, K. Myeloid-derived suppressor cells and plasmacytoid dendritic cells are associated with oncogenesis of oral squamous cell carcinoma. J. Oral Pathol. Med. 2022, 2022, 1–11.
- Chen, I.-C.; Awasthi, D.; Hsu, C.-L.; Song, M.; Chae, C.-S.; Dannenberg, A.J.; Cubillos-Ruiz, J.R. High-Fat Diet–Induced Obesity Alters Dendritic Cell Homeostasis by Enhancing Mitochondrial Fatty Acid Oxidation. J. Immunol. 2022, 209, 69–76.
- Hurley, H.J.; Dewald, H.; Rothkopf, Z.S.; Singh, S.; Jenkins, F.; Deb, P.; De, S.; Barnes, B.J.; Fitzgerald-Bocarsly, P. Frontline Science: AMPK regulates metabolic reprogramming necessary for interferon production in human plasmacytoid dendritic cells. J. Leukoc. Biol. 2020, 109, 299–308.
- Cui, X.; Ye, Z.; Di Wang, D.; Yang, Y.; Jiao, C.; Ma, J.; Tang, N.; Zhang, H. Aryl hydrocarbon receptor activation ameliorates experimental colitis by modulating the tolerogenic dendritic and regulatory T cell formation. Cell Biosci. 2022, 12, 46.
- Oh, J.-S.; Hwang, S.-U.; Noh, K.-E.; Lee, J.-H.; Choi, S.-Y.; Nam, J.-H.; Song, M.-S.; Jung, N.-C.; Song, J.-Y.; Seo, H.G.; et al. Synthetic TGF-β Signaling Agonist-Treated Dendritic Cells Induce Tolerogenicity and Antirheumatic Effects. Curr. Issues Mol. Biol. 2022, 44, 3809–3821.
- Qin, T.; Feng, D.; Zhou, B.; Bai, L.; Yin, Y. Melatonin Suppresses LPS-Induced Oxidative Stress in Dendritic Cells for Inflammatory Regulation via the Nrf2/HO-1 Axis. Antioxidants 2022, 11, 2012.
- Vega, J.A.; García-Suárez, O.; Hannestad, J.; Pérez-Pérez, M.; Germanà, A. Neurotrophins and the immune system. J. Anat. 2003, 203, 1–19.
- Wang, Y.; Fang, J.; Liu, B.; Shao, C.; Shi, Y. Reciprocal regulation of mesenchymal stem cells and immune responses. Cell Stem Cell 2022, 29, 1515–1530.
- Es, H.A.; Bigdeli, B.; Zhand, S.; Aref, A.R.; Thiery, J.P.; Warkiani, M.E. Mesenchymal stem cells induce PD-L1 expression through the secretion of CCL5 in breast cancer cells. J. Cell Physiol. 2020, 236, 3918–3928.
- Chang, J.C.; Chang, H.-S.; Wu, Y.-C.; Cheng, W.-L.; Lin, T.-T.; Chang, H.-J.; Chen, S.-T.; Liu, C.-S. Antitumor Actions of Intratumoral Delivery of Membrane-Fused Mitochondria in a Mouse Model of Triple-Negative Breast Cancers. OncoTargets Ther. 2020, ume 13, 5241–5255.
- Jia, Y.; Zhao, Y.; Zhang, Z.; Shi, L.; Fang, Y.; Chang, C. Aryl hydrocarbon receptor signaling pathway plays important roles in the proliferative and metabolic properties of bone marrow mesenchymal stromal cells. Acta Biochim. Biophys. Sin. 2021, 53, 1428–1439.
- Yuan, B.; Liu, G.; Dai, Z.; Wang, L.; Lin, B.; Zhang, J. CYP1B1: A Novel Molecular Biomarker Predicts Molecular Subtype, Tumor Microenvironment, and Immune Response in 33 Cancers. Cancers 2022, 14, 5641.
- Gribben, J.G.; Ryan, D.P.; Boyajian, R.; Urban, R.G.; Hedley, M.L.; Beach, K.; Nealon, P.; Matulonis, U.; Campos, S.; Gilligan, T.D.; et al. Unexpected Association between Induction of Immunity to the Universal Tumor Antigen CYP1B1 and Response to Next Therapy. Clin. Cancer Res. 2005, 11, 4430–4436.
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 2005, 33, 489–494.
- Kousar, K.; Ahmad, T.; Abduh, M.S.; Kanwal, B.; Shah, S.S.; Naseer, F.; Anjum, S. miRNAs in Regulation of Tumor Micro-environment, Chemotherapy Resistance, Immunotherapy Modulation and miRNA Therapeutics in Cancer. Int. J. Mol. Sci. 2022, 23, 13822.
- Fan, K.; Spassova, I.; Gravemeyer, J.; Ritter, C.; Horny, K.; Lange, A.; Gambichler, T.; Ødum, N.; Schrama, D.; Schadendorf, D.; et al. Merkel cell carcinoma-derived exosome-shuttle miR-375 induces fibroblast polarization by inhibition of RBPJ and p53. Oncogene 2021, 40, 980–996.
- Sun, X.; Lou, L.; Zhong, K.; Wan, L. MicroRNA-451 regulates chemoresistance in renal cell carcinoma by targeting ATF-2 gene. Exp. Biol. Med. 2017, 242, 1299–1305.
- Liu, T.; Zhang, X.; Sha, K.; Liu, X.; Zhang, L.; Wang, B. miR-709 up-regulated in hepatocellular carcinoma, promotes proliferation and invasion by targeting GPC5. Cell Prolif. 2015, 48, 330–337.