Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Major depressive disorder (MDD) is a highly prevalent psychiatric disorder, whose pathophysiology has been linked to the neuroinflammatory process. The increased activity of the Nod-like receptor pyrin containing protein 3 (NLRP3) inflammasome, an intracellular multiprotein complex, is intrinsically implicated in neuroinflammation by promoting the maturation and release of proinflammatory cytokines such as interleukin (IL)-1β and IL-18.
- antidepressants
- bioactive compounds
- depression
- neuroinflammation
1. Introduction
Major depressive disorder (MDD) is a highly debilitating illness of multifactorial etiology that considerably compromises quality of life [1]. There are still several gaps in our current understanding of the pathogenesis and treatment of this disorder, and currently available antidepressants have several limitations [2]. Therefore, a better understanding of the pathophysiology of MDD is of paramount importance in the search for new therapeutic targets.
Over the last decades, it has been proposed that inflammatory processes are involved in the initiation, maintenance and relapse of MDD [3]. Notably, individuals with MDD possess increased levels of proinflammatory mediators, particularly C-reactive protein (CRP), the cytokines interleukin (IL)-6 and IL-1β, and tumor necrosis factor alpha (TNF-α) [4]. In addition, chronic activation of the immune system has been associated with alterations in the function and volume of various brain regions in MDD patients, particularly the hippocampus and medial prefrontal cortex, structures which have been shown to play a role in the regulation of affective behaviors [3]. Furthermore, immune deregulation may promote the dysregulation of several neurotransmitter pathways, including the dopaminergic, serotoninergic, noradrenergic, and glutamatergic systems [3]. Particularly, proinflammatory cytokines can mitigate serotonin synthesis (which has been directly linked to the etiology of MDD), by inducing the enzyme indoleamine 2, 3 dioxygenase (IDO), which catabolizes tryptophan into kynurenine [3].
Noteworthy, the activation of the NLRP3 inflammasome in microglia plays a crucial role during neuroinflammation by synthesizing and releasing IL-1β and IL-18, thus contributing to an increase in inflammatory cytokines [5]. Therefore, neuroinflammatory pathways, and in particular those related to NLRP3, are potential therapeutic targets for the treatment of MDD [2].
2. Neuroinflammation and the NLRP3 Inflammasome
Neuroinflammation is characterized by chronic immune activation originating from a disruption of the communication between brain, microbiota, immune system, and host [6]. The neuroinflammatory process can be triggered by several factors such as stress, gut dysbiosis, infections, neurodegenerative diseases, and stroke [6,7,8]. These factors are capable of initiating an inflammatory response through pathogen-associated molecular patterns (PAMPs) and/or damage-associated molecular patterns (DAMPs), which interact with pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), as well as nucleotide-binding oligomerization domain-like receptors (NLRs) present in microglia [9,10].
Within the neuroinflammatory context, microglia constitute the main mediators of this process, acting as first response cells to endogenous and exogenous insults [5]. Microglia and macrophages are able to differentiate into different phenotypes. Under normal conditions, microglia assume a branched morphology and highly mobile processes for constant monitoring of the brain parenchyma (M2 phenotype). Following an insult, microglia retracts its processes, and adopts an amoeboid form (M1 phenotype) [5,11]. The M1 phenotype positively regulates immune responsive surface proteins, such as major histocompatibility complex type II (MHCII) and chemokine receptors. In addition, it promotes the transcription of genes encoding inflammatory mediators and cytokines [5,12]. A persistent insult promotes microglial reactivation, which culminates in the release of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and IL-18. In addition to cytokine release, this process contributes to the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which in turn induce neurotoxicity [12]. Noteworthy, neuronal damage culminates in the release of ATP and/or ADP, which maintain microglial activation via the P2X purinoreceptor 7 (P2X7) [13,14].
Particularly, inflammasomes play a key role in microglial activation by sensoring various pathogens and cellular derivatives associated with damage and stress [15]. Although several inflammasomes have been described, the best characterized inflammasome related to MDD is the NLR family pyrin domain containing 3 (NLRP3) inflammasome [2,16]. This inflammasome consists of a sensor NLRP3 protein, an adaptor apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) and the effector enzyme caspase-1. The NLRP3 protein is composed of a nucleotide-binding NACHT domain containing ATPase activity, an amino-terminal pyrin (PYD) domain, and a carboxy-terminal leucine-rich repeat (LRR) domain [16].
Activation of NLRP3 in macrophages or microglia may occur by the canonical pathway via two sequential signals: priming and activation [17]. The priming step occurs through a first stimulus promoted by ligands for TLRs, NLRs or cytokine receptors, which induces a morphological alteration into the active phenotype of these cells and promotes the expression of NLRP3 and pro-IL-1β via factor nuclear kappa B (NF-kB) and myeloid differentiation primary response 88 (Myd88) pathways [5,17]. This step also involves post-translational modifications by phosphorylation and ubiquitination of the NLRP3 protein required for inflammasome activation [15,17]. In addition, the initiation step promotes the association of NLRP3, ASC and procaspase-1 to form the inflammasome complex through PYD-PYD interactions between NLRP3 and ASC as well as CARD-CARD (caspase activation and recruitment domains) interactions between ASC and procaspase-1 [18]. Since in this step the proteins form only an inactive NLRP3 oligomeric complex, a second step called activation is required [5]. This second step is triggered by several stimuli that culminate in different cellular and molecular signaling events, such as mitochondrial dysfunction, lysosomal damage, calcium and potassium ion flux, and ROS synthesis [17]. These events activate the NLRP3 inflammasome, leading to autoproteolytic cleavage of procaspase-1 into active caspase-1 [17]. Activated caspase-1, in turn, cleaves pro-IL-18 and pro-IL-1β into the biologically active cytokines IL-18 and IL-1β, respectively [5,19].
In addition to canonical NLRP3 inflammasome activation, a non-canonical pathway may also lead to IL-1β and IL-18 synthesis through NLRP3 inflammasome activation. This may occur through binding of lipopolysaccharide (LPS) to caspases 4/5 in humans or caspase-11 in mice through a process that is independent of TLR [5,17,20]. In addition, monocytes and dendritic cells promote activation of caspase-1 and induce IL-1β secretion, without requiring a secondary stimulus through an alternative pathway [17,21,22]. Once activated, these interleukins undergo exocytosis and bind to their respective receptors IL-1R and IL-18R, which are expressed by glial cells, subsequently inducing the synthesis and release of cytokines and thus leading to an increase in the levels of IL-1β, IL-6 and TNF-α, as seen in MDD [4,5,23,24]. The neuroinflammation-induced increase in IL-1β, TNF-α, and interferon-gamma (IFN-γ) levels positively regulates the IDO enzyme, which in turn increases the conversion of tryptophan into neurotoxic metabolites such as 3-hydroxyquinurenine, 3-hydroxyanthralinic acid and quinolinic acid. Consequently, this results in a decrease in serotonin levels, which is thought to underlie, at least in part, the pathophysiology of MDD [25].
Further to the synthesis and release of cytokines, it has also been shown that the activation of NLRP3 via canonical and non-canonical pathways can lead to gasdermin D-mediated membrane pore formation (GSDMD) and subsequently pyroptosis, characterized by cell swelling and lytic cell death, which culminates in the release of intracellular DAMPs into the extracellular medium [5,18]. A schematic summary of the main events related to the activation of the NLRP3 inflammasome pathway is illustrated in Figure 1.
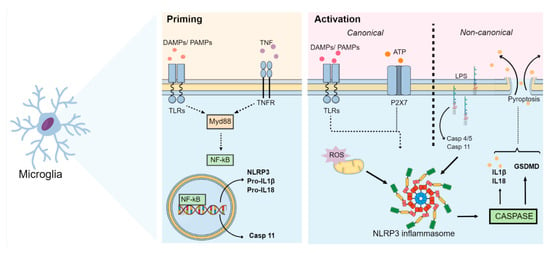
Figure 1. NLRP3 inflammasome pathways. After an insult, the NLRP3 inflammasome initiation step occurs. During this step, DAMPs, PAMPs and cytokines (such as TNF-α) interact with TLRs, NLRs or cytokine receptors (such as TNFR). This interaction induces the activation of the Myd88 and NF-kB pathways. Activated NF-kB then translocates to the nucleus and induces the expression of NLRP3, pro-IL-1β, pro-IL-18 and caspase-11 (through the non-canonical pathway). During this activation step, several stimuli culminate in different cellular and molecular signaling events, such as mitochondrial dysfunction, lysosomal damage, calcium and potassium ion flux, ROS synthesis, and ATP release. These events activate the NLRP3 inflammasome, which cleaves procaspase-1 and releases the active form of caspase-1. Caspase-1 then cleaves pro-IL-18 and pro-IL-1β into their active forms and activates GSDMD, thus inducing pyroptosis. Synthesis of IL-1β and IL-18 as well as activation of GSDMD by NLRP3 may also occur via a non-canonical pathway, in which LPS binds directly to caspases 4/5 in humans and caspase-11 in mice. Abbreviations: DAMPs: damage-associated molecular patterns; GSDMD: gasdermin D-mediated membrane pore formation; IL: interleukin; LPS: lipopolysaccharide; Myd88: myeloid differentiation primary response 88; NF-kB: nuclear factor kappa B; NLR: nucleotide-binding oligomerization domain-like receptor; NLRP3: Nod-like receptor pyrin containing 3; PAMPs: pathogen-associated molecular patterns; TLRs: Toll-like receptors; TNF-α: tumor necrosis factor; TNFR: tumor necrosis factor receptor.
Microglia activation also results in intense astrocytic activation [26]. Chronic activation of these glial cells reduces the synthesis of communicating junctions that form the blood–brain barrier (BBB) and increases the levels of chemokines, such as the C-C motif chemokine ligand 2 (CCL2). This leads to the infiltration of monocytes and macrophages from the circulation into the CNS, contributing to maintain a proinflammatory state [27,28].
3. Involvement of NLRP3 Inflammasome in MDD
Preclinical Evidence
Zhang et al. (2014) conducted the first study to assess inflammasome activation in a mouse model of depression. In this study, lipopolysaccharide (LPS)-treated male BALB/c mice (0.8 mg/kg, i.p.) exhibited an increase in NLRP3 inflammasome mRNA expression, resulting in an increase of proinflammatory cytokine IL-1β mRNA and protein levels in the brain. These changes were accompanied by the induction of depressive-type and anhedonic-type behaviors. In addition, pretreatment with the NLRP3 inflammasome inhibitor Ac-Tyr-Val-Ala-Asp-chloromethylketone (8 mg/kg, i.p.) was capable of mitigating these LPS-induced depressive-like behaviors [42].
In a different study, depressive-like behaviors in male C57BL/6 mice were observed up to 29 days after a single LPS administration (5 mg/kg, i.p.) in a manner dependent on the activation of the NLRP3 inflammasome. It was found that three days after LPS administration, the expression of NLRP3, ASC, caspase-1 p10, TNF-α IL-1β, and IL-18 in the hippocampus and the levels of IL-1β and TNF-α in the serum remained elevated, while hippocampal IL-10 levels decreased as a result of microglial activation [43]. On the other hand, repeated daily administration of increasing doses of LPS (0.1, 0.42 and 0.83 mg/kg; i.p.) has been shown to induce depressive-like behaviors in C57BL/6J mice by a mechanism independent of NLRP3 inflammasome activation [44].
Data from various stress-exposed animal models of depression also suggest that there is a relationship between the development of depressive-like behaviors and the NLRP3 inflammasome [45,46]. Male BALB/c mice subjected to a chronic unpredictable mild stress (CUMS) protocol for 4 weeks displayed depressive-like behaviors and increased hippocampal levels of active IL-1β [46]. In a different study, rats subjected to 6 h of restraint stress for 21 days exhibited hippocampal alterations characterized by increased Iba-1 expression, ROS formation, NF-kB, NLRP3, cleaved caspase-1, IL-1β and IL-18 levels [45].
A study by Pan et al. (2014) observed that male Wistar rats exposed to CUMS for a period of 12-weeks over-expressed P2X7 and TLR2 in the prefrontal cortex, causing activation of the NF-kB pathway and consequently the NLRP3 inflammasome, resulting in microglial activation, astrocytic impairment, and increased prefrontal cortex IL-1β expression [47]. The involvement of the P2X7 receptor in inflammasome activation was further observed in male Sprague Dawley rats submitted to CUMS for 3 weeks. In this study, CUMS induced depressive-like behaviors through a mechanism dependent on the P2X7 receptor. In addition, rats subjected to chronic unpredictable stress (CUS) exhibited increased levels of extracellular ATP, cleaved-caspase 1, ASC, and IL-1β in the hippocampus [13].
Following NLRP3 inflammasome activation, other events including pyroptosis, IDO activation and alterations of the autophagic process have also been associated with the induction of depressive-like behaviors [48,49,50]. Indeed, it has been recently shown that administration of monosodium glutamate results in an increase in the levels of Gasdermin D (GSDMD; a protein that induces pyroptosis), caspase-1, IL-1β, IL-18, NLRP3, and ASC in the hippocampus of postnatal rats [50].
LPS administration (1.8 mg/kg, i.p.) has been shown to increase the expression and activity of IDO in the hippocampus while also inducing depressive-like behaviors in C57BL/6 mice in an NLRP3-dependent manner [51]. Similarly, male Sprague Dawley rats submitted to CUMS for 4 weeks exhibited increased activation of IDO, which caused an increase in the kynurenine/tryptophan ratio and a consequent reduction in serotonin levels. In addition, these animals developed depressive-like behaviors through the activation of the P2X7/NLRP3 inflammasome axis, resulting in increased IL-1β, IL-6, and TNF-α expression [52].
The administration of LPS (500 μg/kg, i.p., every 2 days for a total of seven injections) has been shown to suppress autophagic markers, including LC3II/LC3I and beclin-1, while also inducing neuroinflammation via NLRP3, and this seems to be associated with the occurrence of depressive-like behaviors in rats [37]. Wang et al. (2020) also showed that LPS increased the expression of pro-inflammatory cytokines and caused NLRP3 inflammasome activation paralleled with inhibition of autophagy in the hippocampus of rats and in BV2 cells [53]. In male rats subjected to restraint stress (6-hour for 28 days), the development of depressive-like behaviors was accompanied by a concomitant dysfunction of the AMPK-mTOR (AMP-activated protein kinase-mammalian target of rapamycin) pathway and a decreased expression of LC3-II and beclin-1 in the prefrontal cortex [49]. On the other hand, administration of corticosterone to male mice (20 mg/kg, p.o., for 21 days) was shown to induce the development of depressive-like behaviors without altering autophagy-related proteins phospho-mTORC1, LC3A/B, and beclin-1 in the hippocampus [54].
The microbiota-gut-inflammasome-brain axis is a bidirectional communication system linking psychological stress responses, immune system function, and gut microbiome composition [55,56]. Within this context, it has been suggested that stress-induced increases in NLRP3 signaling can in turn promote pro-inflammatory bacterial clades within the gut microbiota composition. These microbiota changes may alter gut barrier function and result in the increased translocation of bacteria to otherwise sterile enteric compartments, thus contributing to the inflammatory process. Furthermore, dysbiosis can compromise the bioavailability of monoamines and neuroactive compounds, further exacerbating depressive symptoms [55]. In line with this idea, a recent study has shown that transplantation of fecal microbiota from control male Sprague Dawley rats ameliorated the occurrence of depressive-like behaviors in CUMS-exposed rats. Furthermore, fecal microbiota transplantation restored the levels of serotonin and suppressed the activation of microglial and astrocytic cells, reducing the expression of NLRP3, ASC, caspase-1, and IL-1β in the hippocampus of CUS-exposed animals [57,58]. Reinforcing the existence of a link among inflammation, microbiota, and depression, transplantation of fecal microbiota from NLRP3 KO mice has been shown to alleviate chronic stress-induced depressive-like behaviors in recipient mice. In addition, fecal microbiota transplantation also attenuated astrocytic activation [59]. These findings support the notion that stress- or LPS-triggered neuroinflammation can induce depressive-like behaviors by promoting the activation of the NLRP3 inflammasome and that modulation of gut microbiota may be a critical target in alleviating NLRP3-driven neuroinflammation. Figure 2 illustrates the main pathophysiological events underlying the neuroinflammatory process associated with the activation of the NLRP3 inflammasome and the development of depressive-like behaviors in animal models.
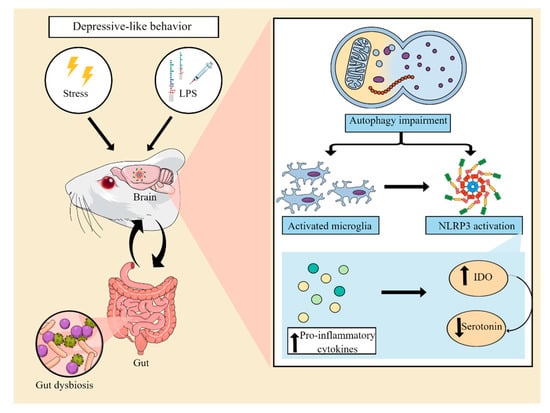
Figure 2. Involvement of the NLRP3 inflammasome in preclinical models of depression. Animal models of depression induced by stress or an inflammatory challenge with lipopolysaccharide (LPS) have been used to elucidate the mechanisms involved in the pathophysiology of MDD. These models are able to induce a neuroinflammatory process, associated with an impairment of autophagic pathways and microglial activation. This process leads to the assembly and activation of the NLRP3 complex. As a consequence, there is an increase in the levels of proinflammatory cytokines, which in turn result in increased indoleamine 2,3 dioxygenase (IDO) activity, decreased tryptophan bioavailability, and a consequent reduction in serotonin synthesis. Furthermore, NLRP3 activation has also been suggested to be associated with gut dysbiosis, compromising the production of serotonin and neuroprotective compounds by the gut microbiota. Abbreviations: NLRP3: Nod-like receptor pyrin containing protein 3.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24010133
This entry is offline, you can click here to edit this entry!