Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
Homozygotyczna hipercholesterolemia rodzinna (HoFH) dotyka średnio 1 na 300 000 osób. Jest to bardzo rzadkie genetyczne zaburzenie metabolizmu lipoprotein. Jest to spowodowane mutacjami w obu allelach genu receptora LDL (LDLR) i rzadziej mutacjami w APOB, ligandzie dla LDLR i konwertazy probiałkowej subtilizyny keksiny typu 9 (PCSK9), białka degradującego LDLR.
- ANGPTL3 inhibitors
- evinacumab
- familial hypercholesterolemia
1. Homozygotyczna rodzinna hipercholesterolemia
Homozygotyczna hipercholesterolemia rodzinna (HoFH) dotyka średnio 1 na 300 000 osób. Jest to bardzo rzadkie genetyczne zaburzenie metabolizmu lipoprotein. Jest to spowodowane mutacjami w obu allelach genu receptora LDL (LDLR), rzadziej mutacjami w APOB, ligandzie dla LDLR i konwertazy probiałkowej subtylizyny keksiny typu 9 (PCSK9), białka degradującego LDLR [ 1 ]. Wyższe poziomy cholesterolu lipoprotein o małej gęstości (LDL-C) charakteryzują się zmianami genetycznymi, które nie wykazują ekspresji receptora LDL (homozygoty zerowe), w porównaniu ze zmianami z dwoma niezerowymi allelami lub jednym zerowym i jednym niezerowym homozygotami, które tylko częściowo zmniejszają aktywność receptora LDL [ 2 , 3]. Mutacje te upośledzają funkcję wątroby polegającą na usuwaniu LDL-C z krwioobiegu, co skutkuje wysokim poziomem cholesterolu całkowitego i LDL-C [ 4 ]. Dla porównania, cząsteczki LDL, które wiążą PCSK9, są celem degradacji i zniszczenia w lizosomach. Mutacje powodujące utratę funkcji genu PCSK9 obniżają poziom LDL-C i zmniejszają ryzyko zawału mięśnia sercowego u osób rasy białej i czarnej oraz zmniejszają ryzyko udaru mózgu u osób rasy czarnej. Można stwierdzić, że inhibitory PCSK9 zapobiegają miażdżycowemu zdarzeniu sercowo-naczyniowemu [ 1 ].
Pacjenci z HoFH od urodzenia mają bardzo wysoki poziom LDL-C, co skutkuje wysokim ryzykiem przedwczesnej miażdżycy i innych chorób sercowo-naczyniowych. Klinicznie HoFH charakteryzuje się stężeniem LDL-C > 500 mg/dl (>13 mmol/l). Statyny i leki hipolipemizujące są w dużej mierze zależne od aktywności receptora LDL; dlatego u pacjentów z dwoma allelami zerowymi mogą wykazywać zmniejszoną skuteczność. Dlatego większość pacjentów z HoFH nie osiąga zalecanych w wytycznych poziomów LDL-C pomimo leczenia wieloma lekami [ 3 ]. Niestety, mutacje u pacjentów z hipercholesterolemią rodzinną wiążą się ze zwiększonym (nawet 3,8-krotnie) prawdopodobieństwem wystąpienia zawału mięśnia sercowego w wieku poniżej 55 lat [ 5 ].]. Wczesne rozpoznanie FH i obserwacja, z kompleksową opieką podłużną, ze szczególnym uwzględnieniem niedrożności i zwężenia zastawki aortalnej, mają kluczowe znaczenie w zapobieganiu przedwczesnej miażdżycowej chorobie sercowo-naczyniowej (ASCVD) [ 2 ].
Zgodnie z wytycznymi European Society of Cardiology (ESC) i European Society of Atherosclerosis (EAS) z 2019 r. poziom LDL-C powinien być niższy niż 1,42 mmol/L (55 mg/dL) u pacjentów z bardzo dużym ryzykiem ASCVD, poniżej 1,81 mmol/l (70 mg/dl) u pacjentów z grupy wysokiego ryzyka i poniżej 2,59 mmol/l (100 mg/dl) u pacjentów z grupy umiarkowanego ryzyka [ 5 ].
2. Układ białek ANGPTL3, 4 i 8 – charakterystyka i rola w metabolizmie lipidów
Białka podobne do angiopoetyn (ANGPTL) to rodzina białek składająca się z członków 1–8 angiopoetyn, które różnią się pod względem ekspresji tkankowej i regulacji. Każdy z nich składa się ze wspólnej domeny na końcu aminowym (N-koniec), domeny typu coiled-coil (CCD), domeny podobnej do fibrynogenu (FLD) na C-końcu grupy karboksylowej i regionu łącznikowego. Angiopoetyna-8 różni się od innych ANGPTL tym, że nie zawiera domeny podobnej do fibrynogenu na C-końcu [ 6 ]. Białka ANGPTL należą do rodziny czynników wzrostu śródbłonka naczyniowego (VEGF) i odgrywają różne role w procesach biologicznych i patologicznych, w tym regulacji hormonalnej, metabolizmie glukozy i insulinooporności [ 7 ].
ANGPTL3, ANGPTL4 i ANGPTL8 są najważniejsze w metabolizmie lipoprotein, ponieważ są odpowiedzialne za metabolizm trójglicerydów (TG) - bogatych w triglicerydy lipoprotein (chylomikrony, VLDL) - poprzez hamowanie aktywności lipazy lipoproteinowej (LPL), VLDL i LDL za pośrednictwem hamowanie lipazy śródbłonkowej (EL) [ 6 , 8 ]. Aktywność LPL jest zmniejszana poprzez zmianę konformacji z homodimerycznej, która jest biologicznie aktywna, na biologicznie nieaktywną lub monomeryczną. LPL to enzym wytwarzany w komórkach tłuszczowych i mięśniowych, który ogranicza szybkość hydrolizy lipoprotein bogatych w TG do wolnych kwasów tłuszczowych (FFA). Gdy ten proces jest zaburzony, w osoczu dochodzi do ciężkiej hipertriglicerydemii [ 9]. Najbardziej znanym ANGPTL jest ANGPTL3, który został odkryty w 1999 roku. ANGPTL3 jest produkowany w wątrobie. W następnym roku, 2000, odkryto ANGPTL4, który jest wytwarzany w wątrobie, mięśniach szkieletowych, tkance tłuszczowej, jelitach, mózgu i sercu. Dodatkowo ANGPTL8 został odkryty w 2012 roku, a jego głównym źródłem jest tkanka tłuszczowa i wątroba [ 10 ].
ANGPTL3, 4 i 8 kontrolują dostępność lipoprotein bogatych w triglicerydy, LDL i cholesterolu lipoprotein o dużej gęstości (HDL-C), w zależności od stanu odżywienia organizmu, temperatury i aktywności fizycznej, poprzez regulację wydzielania LPL. Aktywność LPL wzrasta po posiłku, a triglicerydy są magazynowane w białej tkance tłuszczowej WAT. Natomiast po posiłku aktywność LPL jest zmniejszona w sercu, brązowej tkance tłuszczowej i mięśniach szkieletowych przez ANGPTL3 i 8 (ekspresja ANGPTL8 jest szczególnie zwiększona). Odwrotna sytuacja ma miejsce podczas postu, kiedy aktywność LPL wzrasta w sercu, brunatnej tkance tłuszczowej i mięśniach szkieletowych. W białej tkance tłuszczowej aktywność LPL na czczo jest zmniejszana przez ANGPTL4 [ 11 , 12 , 13 , 14 ,15 ] ( Rysunek 1 ).
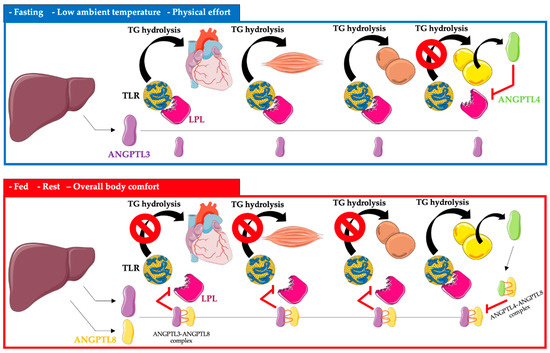
Rycina 1. Regulacja metabolizmu trójglicerydów w sercu, mięśniach, brązowej i białej tkance tłuszczowej przez ANGPTL3, ANGPTL4 i ANGPTL8. Skróty: TG – trigliceryd; TLR – lipoproteina bogata w triglicerydy; LPL – lipaza lipoproteinowa; ANGPTL3 — białko podobne do angiopoetyny 3; ANGPTL4 — białko podobne do angiopoetyny 4; ANGPTL8 — białko podobne do angiopoetyny 8. Do przygotowania rysunku wykorzystano: https://smart.servier.com (bezpłatny dostęp; 20 października 2022 r.).
ANGPTL3 oprócz wpływu na LPL zmniejsza również aktywność EL, co prowadzi do spowolnienia metabolizmu lipoprotein bogatych w triglicerydy [ 16 ].
3. ANGPTL3, 4 i 8 jako biomarkery ryzyka sercowo-naczyniowego
Kilka publikacji donosi, że niedobór ANGPTL3 chroni przed chorobą wieńcową (CAD). Według badań Stitziela i wsp. u osób z całkowitym niedoborem ANGPTL3 w tętnicach wieńcowych brakowało blaszki miażdżycowej [ 12 ]. Ponadto zdrowi pacjenci wykazywali niższe stężenia ANGPTL3 w porównaniu z pacjentami, którzy doświadczyli zawału mięśnia sercowego (MI) [ 12 ]. U pacjentów ze stężeniem ANGPTL33 18–271 ng/ml ryzyko zawału serca zmniejszało się nawet o 29%. Naukowcy wykazali również związek mutacji powodującej utratę funkcji (LOF) w ANGPTL3 z ryzykiem CAD. Poziomy lipoprotein o małej gęstości LDL-C, HDL-C o dużej gęstości i TG zależą od LOF w ANGPTL3. Pacjenci z mutacją LOF wykazywali 34% zmniejszenie ryzyka CAD w porównaniu z pacjentami, którzy nie byli nosicielami mutacji LOF. Ponadto pacjenci z mutacją LOF wykazywali o 11% niższy poziom cholesterolu całkowitego, 12% niższy poziom LDL i 17% niższy poziom TG w porównaniu z pacjentami bez mutacji. Oprócz faktu, że utrata ANGPTL3 zwiększa aktywność LPL, prowadząc do zmniejszenia TG i lipoprotein bogatych w LDL, może wpływać na wrażliwość na insulinę i odgrywać ważną rolę w hemostazie glukozy [ 12 ].
W innym badaniu wpływu ANGPTL3 i 4 na CAD zespół Sun i in. przedstawił wyniki badania z udziałem 305 pacjentów. Wysoki poziom ANGPTL3 był ściśle związany z ciężkością zmian miażdżycowych w naczyniach wieńcowych, natomiast poziom ANGPTL4 był obniżony. Poziomy tych glikoprotein mogą mieć znaczący wpływ na rozwój CAD [ 17 ]. W innym badaniu wykazano związek między mutacjami inaktywującymi gen ANGPTL4 a ryzykiem wystąpienia choroby niedokrwiennej serca. Badanie to obejmowało ponad 42 000 osób. Dewey i in. w 2017 roku udowodnili, że obniżenie poziomu TG, cholesterolu całkowitego i LDL-C było spowodowane inaktywacją ANGPTL4 przez heterozygotyczną mutację E40k. Pacjenci z tą mutacją ANGPTL4 wykazywali o 19% mniejsze ryzyko choroby niedokrwiennej serca [18 ]. W podobnym badaniu prowadzonym przez Stitziela i wsp. pacjenci z mutacją E40K ANGPTL4 wykazywali o około 35% niższe stężenie TG. Dodatkowo ryzyko choroby niedokrwiennej serca było o 53% niższe. Jednak nie zaobserwowano znaczącego wpływu ANGPTL4 p.E40K na LDL-C [ 19 ]. Zespół badawczy Gusarovej i in. wykazali wpływ mutacji E40K ANGPTL4 na zmniejszenie ryzyka cukrzycy typu 2 o 12%. Badanie to przeprowadzono na 58 000 uczestników badania DiscovEHR [ 20]. Podobne wyniki przedstawił zespół Klarin et al. w badaniu 310 000 osób. Oceniono wpływ utraty funkcji (LOF) mutacji ANGPTL4 na ryzyko choroby niedokrwiennej serca i cukrzycy typu 2. Wykazano, że ryzyko choroby niedokrwiennej serca zmniejszyło się o 16%, a ryzyko cukrzycy typu 2 o 12% [ 21 ].
4. Struktura ewinakumabu i mechanizm działania
Ewinakumab (Evkeeza ® ; dawniej RENG1500) to w pełni ludzkie przeciwciało monoklonalne, hamujące krążący ANGPTL3, które zostało wynalezione przez firmę Regeneron Pharmaceuticals Inc. [ 3 ] i wyprodukowane metodą hodowli komórkowej z genetycznie zmodyfikowanymi rekombinowanymi komórkami jajnika chomika chińskiego [ 22 ]. Ewinakumab jest przeciwciałem monoklonalnym IgG4 składającym się z dwóch ludzkich łańcuchów ciężkich (453 aminokwasy każdy) połączonych wiązaniami dwusiarczkowymi i ludzkich łańcuchów lekkich kappa (214 aminokwasów). Łańcuchy ciężkie są kowalencyjnie połączone wiązaniami dwusiarczkowymi z łańcuchami lekkimi [ 22 ].
Ewinakumab został zatwierdzony przez amerykańską Agencję ds. Żywności i Leków w lutym 2021 r. oraz Europejską Agencję Leków (EMA) w czerwcu 2021 r. i jest obecnie dostępny na rynku pod nazwą handlową Evkeeza do leczenia pacjentów dorosłych i młodzieży (≥12 lat) z homozygotyczną rodzinną hipercholesterolemia [ 23 ]. Zalecana dawka tego nowego leku wynosi 15 mg/kg, podawana w infuzji dożylnej (IV) przez godzinę raz w miesiącu [ 23 ].
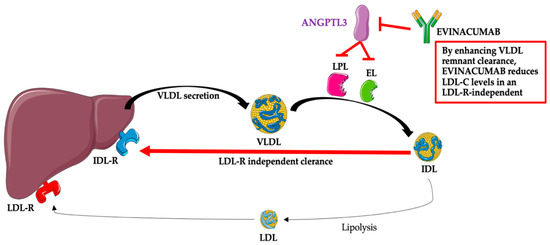
Po podaniu ewinakumab wiąże swój cel, ANGPTL3, i hamuje jego funkcję, co prowadzi do zwiększenia aktywności LPL i EL oraz obniżenia stężenia TG, LDL-C i HDL-C w osoczu [ 24 ]. Mechanizm związany z redukcją LDL-C przez ewinakumab nie jest w pełni poznany; jednakże efekt ten jest niezależny od receptora LDL, a zatem prawdopodobnie wynika z promowania przetwarzania lipoprotein o bardzo małej gęstości (VLDL) i klirensu formowania się LDL w górę [ 12 , 13 , 14 , 15 , 24 ]. Mechanizm działania ewinakumabu przedstawiono na rycinie 2 .
Rycina 2. Ewinakumab — mechanizm działania. Skróty: ANGPTL3 — białko podobne do angiopoetyny 3; EL – lipaza śródbłonkowa; IDL — lipoproteina o średniej gęstości (pozostałości VLDL); IDL-R — receptor lipoprotein o średniej gęstości (remnant receptor VLDL); LDL — lipoproteiny o małej gęstości; LDL-C — lipoproteina o małej gęstości; LDL-R — receptor lipoprotein o małej gęstości; LPL – lipaza lipoproteinowa; VLDL — lipoproteina o bardzo małej gęstości. Do przygotowania rysunku wykorzystano: https://smart.servier.com (bezpłatny dostęp; 20 października 2022 r.).
This entry is adapted from the peer-reviewed paper 10.3390/jcm12010168
This entry is offline, you can click here to edit this entry!