Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The mechanistic target of rapamycin (mTOR) complex 1, mTORC1, integrates nutrient and growth factor signals with cellular responses and plays critical roles in regulating cell growth, proliferation, and lifespan. mTORC1 signaling has been reported as a central regulator of autophagy by modulating almost all aspects of the autophagic process, including initiation, expansion, and termination. An increasing number of studies suggest that mTORC1 and autophagy are critical for the physiological function of skeletal muscle and are involved in diverse muscle diseases.
- mTORC1
- autophagy
- muscle diseases
1. Introduction of mTORC1
The mechanistic target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase. It was first identified as a target protein of rapamycin through the screening of rapamycin-resistant mutants in yeast [1]. Rapamycin can form a complex with FK506-binding protein (FKBP) that inhibits cell cycle progression by inhibition of the yeast TOR1 and TOR2 [1]. Subsequent studies in mammalian cells identified the homologous protein, termed mTOR, which shares more than 40% conservation in amino acid sequence with yeast TOR1 and TOR2 [2]. mTOR exists as two structurally and functionally different complexes, known as mTOR complex 1 (mTORC1) and 2 (mTORC2) respectively (Figure 1). mTORC1 is composed of the central mTOR kinase, the scaffolding protein LST8 [3], the inhibitory subunit DEPTOR [4], the Tti1/Tel2 complex required for mTOR complex stability and assembly [5], Raptor, and PRAS40. Raptor (regulatory-associated protein of mTOR) is critical for mTORC1 assembly, stability, and substrate specificity [6][7], and associates with PRAS40 (Proline-rich AKT substrate 40 kDa), which functions as an inhibitor of mTORC1 activity [8]. mTORC2 shares the components mTOR, LST8, DEPTOR and Tti1/Tel2 that are similarly present in mTORC1, in addition to the unique subunits, Rictor and SIN1 [9]. mTORC1 and mTORC2 can be distinguished by their acute response to rapamycin [10]. Rapamycin is an antifungal metabolite produced by Streptomyces hygroscopicus, which possess immunosuppressive and anti-proliferative properties in mammalian cells. Rapamycin allosterically inhibits mTORC1. In contrast, mTORC2 shows insensitivity to acute rapamycin treatment [10][11].
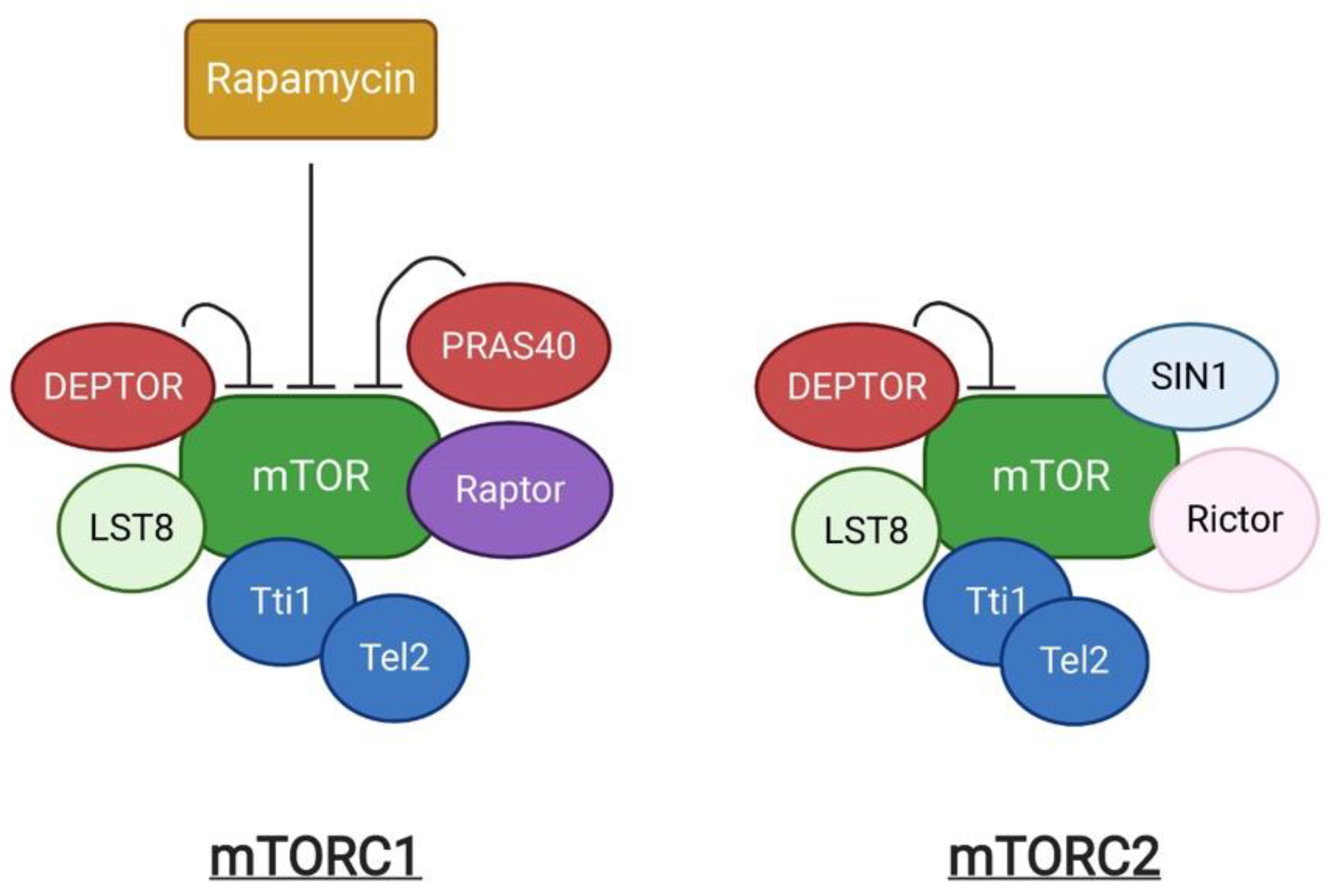
Figure 1. A schematic representation of mTOR complexes.
Genetic studies and pharmacological research using rapamycin have demonstrated that mTORC1 is a master regulator of cell growth and metabolism which senses and integrates different nutritional and environmental cues, including growth factors, energy levels, cellular stress, and amino acids [12]. Growth factor activation of mTORC1 is mediated by a Ras family GTPase, RAS homolog enriched in brain (Rheb), following receptor-coupled PI3K activation and the AKT-dependent phosphorylation and inhibition of the tuberous sclerosis complex (TSC) 1/2 [13]. The TSC complex can also be stabilized by LKB1-AMPK signaling [14]. In contrast, amino acids can stimulate mTORC1 in a PI3K/Akt axis-independent manner and induce the translocation of mTORC1 to the lysosomal surface where mTORC1 is activated upon contact with Rheb. This process requires many complexes, such as the v-ATPase, Ragulator, the Rag GTPases, and GATOR1/2 [15].
2. Regulation of Autophagy by mTORC1
In the absence of nutrients or growth factors, mTORC1 activity is reduced. In turn, it activates cellular catabolic processes while suppressing anabolic processes. Reduced mTORC1 activity increases the activity of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and inhibits ribosomal protein S6 kinase 1 (S6K1) to block protein translation [16]. mTORC1 inactivation also activates autophagy, an evolutionarily conserved catabolic process that recycles long-lived proteins or damaged organelles and provides energy or macromolecular precursors for cell survival [17][18]. This degradation process is coordinately modulated by multiple autophagic regulators [19][20][21][22][23]. Among these regulators, mTORC1 signaling plays critical roles in regulating each step of the autophagy process, including induction, nucleation, elongation, and formation of a double-membrane autophagosome, followed by the fusion of the autophagosome with a lysosome to form autolysosomes to degrade and recycle autophagosome-sequestered substrates (Figure 2).
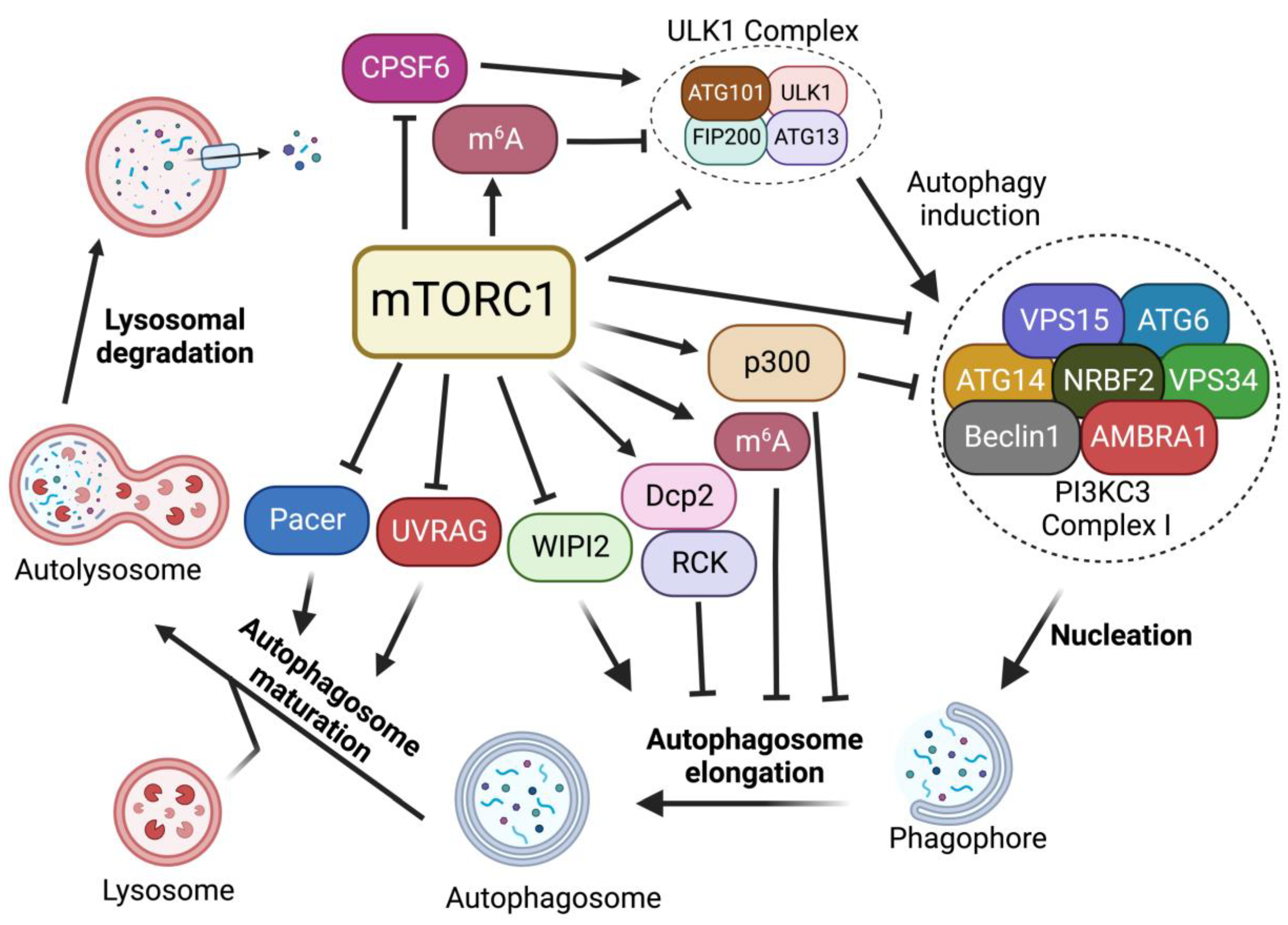
Figure 2. Regulation of autophagy by mTORC1. mTORC1 plays critical roles in the regulation of each step of the autophagy procedure, including autophagy initiation, nucleation, membrane expansion, and termination.
2.1. Regulation of ULK1 Complex during Autophagy Initiation
In mammals, the activation of Unc51-like kinase 1 (ULK1) complex, composed of the Ser/Thr kinase ULK1, ATG13, FIP200, and ATG101, is essential for autophagy induction. The ULK1 complex serves as the main interface where the direct intervention of mTORC1 in autophagy occurs and its activity is mainly regulated by mTORC1- and AMP-activated protein kinase (AMPK)-mediated phosphorylation [24][25]. In nutrient-rich conditions where mTORC1 is active, mTORC1 phosphorylates ULK1 at the Ser757 site, disrupting the interaction of AMPK and ULK1, leading to the inhibition of autophagy induction [24]. mTORC1 also phosphorylates ATG13, a component of ULK1 complex, at Ser258 to decrease ULK1 complex activity and suppress autophagy [26]. During starvation and environmental stresses where mTORC1 is inactive, mTORC1 dissociates from the ULK1 complex and ULK1 is activated by AMPK-dependent phosphorylation at multiple sites [24]. ULK1 phosphorylation by AMPK is able to induce autophagy [24]. In addition, recent studies suggest that mTORC1-mediated autophagy via ULK1 can also be regulated by protein kinase G 1 (PKG1) in the heart [27][28]. Together, these findings suggest that mTORC1 plays critical roles in autophagy induction through the regulation of the ULK1 complex.
2.2. Regulation of PI3KC3-Beclin 1-ATG14 Complex during Nucleation
After ULK1-mediated autophagy initiation, the nucleation of phagophore requires the lipid kinase activity of class III phosphatidylinositol 3-kinase (PI3KC3), also known as Vacuolar protein sorting 34 (Vps34), which generates phosphatidylinositol-3 phosphate (PI3P) from phosphatidylinositol at the phagophore to promote pre-autophagosome formation. PI3KC3 forms two distinct complexes (PI3KC3-CI and PI3KC3-CII). PI3KC3, VPS15, and Beclin 1 make up the core subunits that are conserved in both complexes. Complex I (PI3KC3-CI) contains ATG14, activating molecule in Beclin 1-regulated autophagy protein 1 (AMBRA1) and nuclear receptor-binding factor 2 (NRBF2), while complex II includes UV radiation resistance-associated gene protein (UVRAG) [29]. Among them, PI3KC3-CI has been shown to regulate the nucleation of phagophores. Upon mTORC1 inhibition or ULK1 complex activation, ULK1 complex targets the sub-domains of the endoplasmic reticulum (ER), known as omegasomes, and recruits PI3KC3-CI to produce PI3P at phagophore, facilitating the nucleation of autophagosomes [30][31].
mTORC1 inhibition activates the ULK1 complex, which in turn induces the phosphorylation of Beclin 1 at Ser15 and Ser30, thus causing the activation of the PI3KC3-CI [32][33]. In addition, it has been reported that mTORC1 can also directly regulate the activity of the PI3KC3-CI by phosphorylating its components ATG14, AMBRA1, and NRBF2. mTORC1 inhibits PI3KC3 activity by directly phosphorylating ATG14 on multiple sites. Mutation of these phosphorylation sites on ATG14 enhances autophagic flux [34]. NRBF2 can be phosphorylated by mTORC1 at S113 and S120. The inhibition of such phosphorylation events increases VPS34 complex assembly and activity, enhancing autophagy flux [35]. In addition, mTORC1 can directly phosphorylate AMBRA1 at Ser52 under normal conditions. The inhibition of mTORC1 results in the dephosphorylation of AMBRA1 and increases its interaction with E3-ligase TRAF6, which subsequently ubiquitinates ULK1 on Lys-63. This K63-linked ubiquitination stabilizes ULK1 and induces its kinase activity, leading to an increase in autophagy induction [30]. Thus, these studies suggest that mTORC1 strictly restricts the nucleation of phagophores through the targeting of multiple components of the PI3KC3-CI complex.
2.3. Regulation of Autophagosome Expansion
When PI3P is generated at the omegasome, PI3P subsequently recruits PtdIns3P-binding effectors, including Atg18/WD-repeat domain phosphoinositide-interacting protein-2 (WIPI-2) and FYVE domain-containing protein 1 (DFCP1), which in turn recruit more autophagy machinery proteins [36][37]. WIPI2 binds to ATG16L and recruits the ATG12-ATG5-ATG16L complex to the phagophore. The WIPI-2-recruited ATG12-ATG5-ATG16L complex is part of a conjugation system where the E1-like ATG7 transfers LC3-I to the E2-like enzyme ATG3 that associates with ATG12 on the complex, catalyzing the conjugation of ATG8/LC3 proteins with membrane resident phosphatidylethanolamine (PE) [38]. This lipidation of the ATG8/LC3 process generates LC3-II, which is the characteristic signature of autophagic membranes and is involved in the ATG9 vesicle sequestration of cargo [39][40].
Multiple studies have demonstrated that mTORC1 negatively regulates autophagosome expansion by phosphorylating WIPI2 and p300 acetyltransferase [41][42]. mTORC1 phosphorylates WIPI2 at Ser395 and the phosphorylated WIPI2 interacts with E3 ligase HUWE1 to promote its proteasomal degradation. The mTORC1-regulated protein stability of WIPI2 affects both basal and starvation-induced autophagy [41]. Moreover, mTORC1 also directly phosphorylates p300 acetyltransferase to activate p300 by relieving its autoinhibition. P300-mediated LC3 acetylation prevents its interaction with E1 ubiquitin ligase ATG7, thus leading to autophagy inhibition. Upon starvation, reduced mTORC1 activity induces the dephosphorylation of p300. This dephosphorylation event inactivates p300 and causes the deacetylation of LC3, thereby increasing LC3-ATG7 interaction, LC3 lipidation, and autophagosome expansion [42].
2.4. Regulation of Autophagosome Maturation and Termination
After becoming fully sealed, the autophagosome subsequently fuses with lysosome to degrade the engulfed material. Autophagosomes and lysosomes are tethered by diverse tethering machinery, such as Rab GTPases, homotypic fusion and vacuole protein sorting (HOPS) complex, ATG14, and UVRAG [43][44][45]. The fusion of the autophagosome membrane with the lysosome membrane involves the HOPS complex that tethers autophagosomes to lysosomes as well as Syntaxin-17 (Stx-17) to facilitate membrane fusion [46]. Pacer was identified as a vertebrate-specific autophagy activator and is found to facilitate the biogenesis of PI3P on autophagosomes. Furthermore, Pacer interacts with Stx17 and recruits HOPS to autophagosomes [47]. In addition, UVRAG, a component of the PI3KC3 complex II (PI3KC3-CII), is required for the interaction between the autophagosome and PI3KC3-CII. The association of UVRAG with HOPS increases autophagosome-lysosome fusion [44]. At the last stage of autophagy, lysosomal membranes are recycled from autolysosomes to maintain lysosome homeostasis, a process termed Autophagic lysosome reformation (ALR) [48].
It has been shown that mTORC1 directly interacts with and phosphorylates UVRAG at Ser498 in nutrient-rich conditions [49]. This mTORC1-dependent phosphorylation of UVRAG inhibits the interaction between UVRAG and the HOPS complex. However, upon starvation, the dephosphorylation of UVRAG induced by mTORC1 inactivation promotes its interaction with HOPS and enhances autophagosome maturation [49]. Besides Ser498, mTORC1 also phosphorylates UVRAG at Ser550 and Ser571 to maintain ALR [50]. The phosphorylation of UVRAG at Ser550 and Ser571 induces the activation of PI3KC3 and generates PI3P at lysosome. The mutation of these mTORC1-dependent phosphorylation sites on UVRAG causes the failure of lysosome regeneration and induces cell death during starvation, suggesting the indispensable function of mTOR in ALR [50]. A recent study has identified Pacer as a substrate of mTORC1. mTORC1 phosphorylates Pacer at Ser157 and this phosphorylation abolishes the interaction of Pacer with HOPS and Stx17, thus preventing autophagosome maturation during nutrient-rich conditions [51]. During starvation, dephosphorylated Pacer in turn recruits HOPS complex for autophagosome maturation [51].
2.5. mTORC1-Dependent RNA Metabolism
While earlier studies mostly focused on mTORC1 downstream signals linked to cytoplasmic protein metabolism, recent studies have shown that mTORC1 signaling plays critical roles in RNA metabolism, ranging from pre-mRNA splicing, polyadenylation, and mRNA methylation. mTORC1 activation has been shown to induce global mRNA 3′-UTR shortening [52]. In the previous study, researchers further found that mTORC1 regulates RNA processing of autophagy-related gene (Atg) transcripts and alters ATG protein levels and activities through the cleavage and polyadenylation (CPA) complex. Specifically, mTORC1 activity suppresses CDK8 and DOA/CLK2 kinases, which directly phosphorylate CPSF6, a component of the CPA complex. The phosphorylation status of CPSF6 affects its localization, RNA binding, and starvation-induced alternative RNA processing of Atg1/ULK1 and Atg8/LC3 transcripts, nutrient, and energy metabolism, thereby modulating autophagy and metabolism [53]. Alternatively, mTORC1 also regulates the phosphorylation of the decapping enzyme Dcp2. Phosphorylated Dcp2 associates with RCK family members and binds to Atg8/LC3 transcripts to degrade them, leading to autophagy inhibition [54]. Furthermore, recent studies further demonstrate the roles of mTORC1 in RNA methylation. mTORC1-activated S6K enhances the translation of Wilms’ tumor 1-associated protein (WTAP), an adaptor for the N6-methyladenosine (m6A) RNA methyltransferase complex, leading to an increase in m6A levels. Increased m6A activity promotes c-Myc transcriptional activity and the proliferation of mTORC1-activated cancer cells [55]. In addition, mTORC1 stabilizes the m6A methyltransferase complex through the chaperonin containing TCP1 (CCT) complex. The upregulation of m6A modification promotes the degradation of Atg transcripts, including Atg1 and Atg8, and inhibits autophagy [56]. Collectively, these studies uncover another layer of mTORC1 regulation of autophagy through RNA metabolisms.
3. Roles of the mTORC1-Autophagy Pathway in Regulating Skeletal Muscle Functions
Skeletal muscle is the most abundant tissue, comprising ~40% of body mass in humans, and plays key roles in locomotion and maintaining metabolism. It serves as a protein reservoir in the human body which undergoes rapid turnover, a process strictly controlled by the balance between protein synthesis and degradation. As a result, skeletal muscle possesses an extreme sensitivity to the changes in both autophagy and mTORC1 activities. To date, excessive muscle loss has been used as a prognostic index of negative outcomes for a variety of diseases ranging from cancer, infections, and unhealthy aging [57].
3.1. mTORC1, but Not mTORC2, Regulates Skeletal Muscle Sizes
In response to exercise or hormonal stimulation, new proteins are generated, increasing cellular volume and muscle growth, a process named hypertrophy. In contrast, catabolic conditions such as cancer, infections, diabetes, aging, or inactivity/disuse promote a net loss of proteins, causing shrinkage of the muscle volume, a condition named atrophy [58]. Therefore, the balance between biogenesis versus destruction defines the size and the function of muscle cells.
Altered mTOR activity has been linked to both muscle hypertrophy and atrophy. Muscle-specific mTOR knockout mice exhibit severe myopathy leading to premature death between 22 and 38 weeks of age [59]. The mTOR and Raptor (the scaffold protein of mTORC1) knockout mice exhibited decreased postnatal growth due to the reduced size of fast-twitch muscles and displayed a progressive muscle atrophy phenotype [59][60]. However, muscle-specific Rictor (a component of mTORC2) knockout mice fail to show any significant phenotype. Similarly, a recent study reported that muscle specific mTOR and Raptor double knockout in mice induces muscle atrophy and a slower muscle relaxation, which may be caused by a shift of muscle types, from fast-twitch fibers to slow-twitch fibers, and changes in the expression levels of calcium-related genes. The double knockout mice exhibit more severe phenotypes compared to the mice with the deletion of either Raptor or mTOR alone [61]. Consistent with these results, the treatment of rapamycin, an inhibitor of mTORC1, suppresses muscle growth [62]. The deletion of muscular S6K1, a mTORC1 downstream target, induces energy stress and muscle cell atrophy [63]. These studies demonstrate that mTORC1, not mTORC2, is the major regulator in the control of muscle fiber size.
3.2. The Roles of mTORC1-Autophagy Axis in Muscle Homeostasis
Interestingly, an early study reported that acute activation of mTORC1 in vivo drives muscle hypertrophy in the short-term [64]; however, chronic mTORC1 activation by TSC1 depletion in the muscle leads to severe and progressive muscle atrophy, along with low body mass and early death [65]. A recent study also observed that activation of mTORC1 in aged muscle leads to progressive muscle fiber damage, fiber death, and loss of muscle mass [66]. These muscle atrophy phenotypes induced by mTORC1 hyperactivation are primarily due to the lack of autophagy in muscles [65]. Autophagy provides energy and building blocks for metabolisms and thus regulates the level of amino acids, lipids, carbohydrates, and nucleic acids [67][68]. It is also required for intracellular quality control. The inhibition of autophagy leads to the accumulation of ubiquitinated protein aggregates and inclusion bodies as well as cause abnormalities in mitochondria, peroxisomes, ER, and Golgi. For instance, similar to the phenotypes in TSC1-deficient mice, the muscle-specific deletion of Atg7 causes muscle atrophy and muscle force decreases [69]. The accumulation of abnormal mitochondria, sarcoplasmic reticulum distension, the disorganization of sarcomere, and the formation of aberrant concentric membranous structures were observed in Atg7-null muscles, showing that autophagy is essential to preserving muscle mass and maintaining muscle integrity [69]. Therefore, these results suggest that both chronically aberrant increases and decreases in mTORC1 activity and deregulated autophagy result in muscle atrophy, implying that dynamic synthesis–degradation oscillations modulated by the balance of the mTORC1-autophagy axis are essential for maintaining muscle homeostasis (Figure 3).
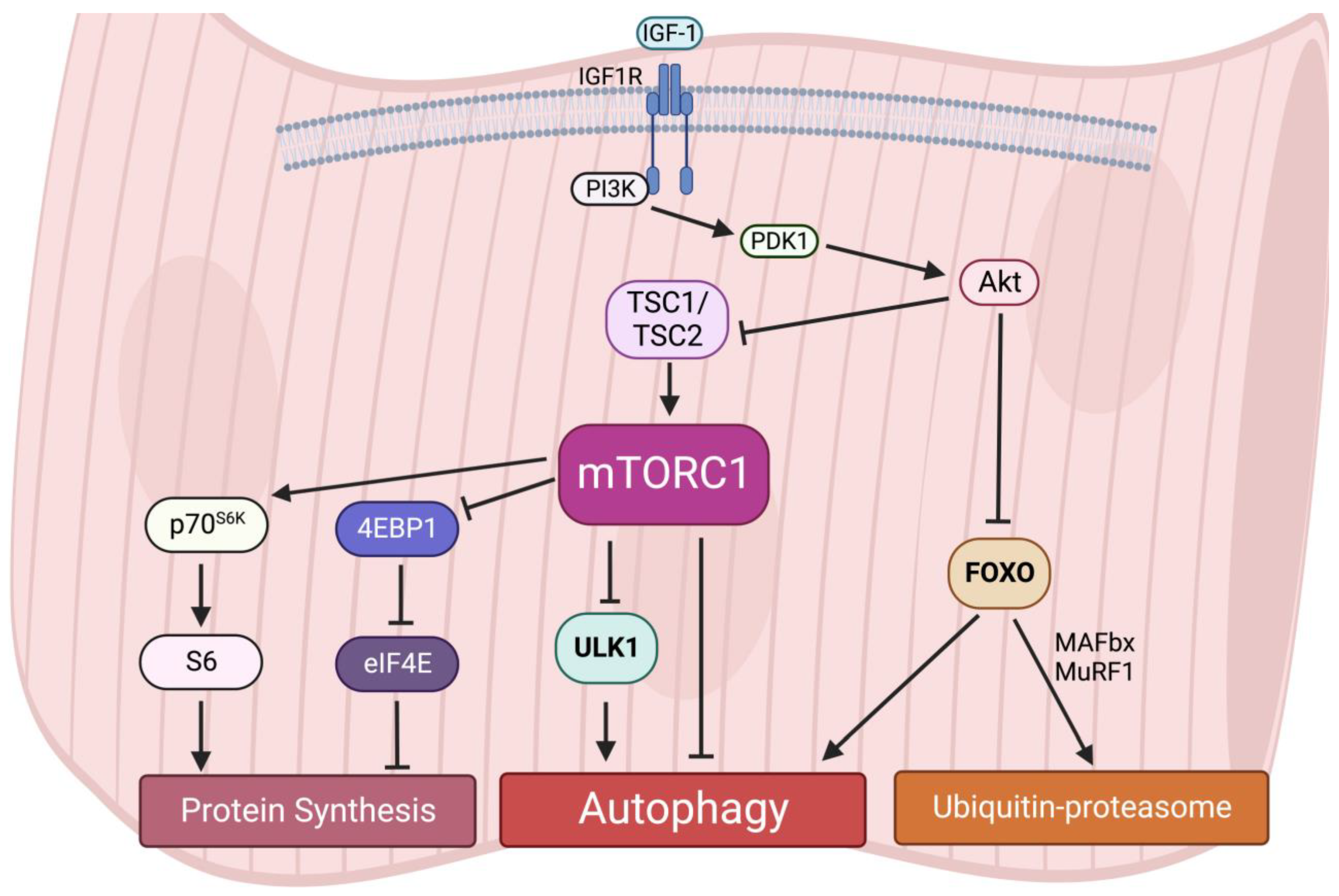
Figure 3. Protein homeostasis is regulated by the mTORC1-autophagy axis in muscles.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24010297
References
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909.
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822.
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904.
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886.
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116.
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464.
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175.
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044.
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201.
- Blenis, J. TOR, the Gateway to Cellular Metabolism, Cell Growth, and Disease. Cell 2017, 171, 10–13.
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371.
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203.
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555.
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575.
- Shimobayashi, M.; Hall, M.N. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016, 26, 7–20.
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494.
- Deleyto-Seldas, N.; Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731.
- Wang, W.; Li, J.; Tan, J.; Wang, M.; Yang, J.; Zhang, Z.M.; Li, C.; Basnakian, A.G.; Tang, H.W.; Perrimon, N.; et al. Endonuclease G promotes autophagy by suppressing mTOR signaling and activating the DNA damage response. Nat. Commun. 2021, 12, 476.
- Tang, H.W.; Wang, Y.B.; Wang, S.L.; Wu, M.H.; Lin, S.Y.; Chen, G.C. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011, 30, 636–651.
- Tang, H.W.; Liao, H.M.; Peng, W.H.; Lin, H.R.; Chen, C.H.; Chen, G.C. Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev. Cell 2013, 27, 489–503.
- Tang, H.-W.; Spirohn, K.; Hu, Y.; Hao, T.; Kovács, I.A.; Gao, Y.; Binari, R.; Yang-Zhou, D.; Wan, K.H.; Bader, J.S.; et al. Next-generation large-scale binary protein interaction network for Drosophila. bioRxiv 2022.
- Chen, G.C.; Lee, J.Y.; Tang, H.W.; Debnath, J.; Thomas, S.M.; Settleman, J. Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy 2008, 4, 37–45.
- Wang, Y.; Goh, K.Y.; Chen, Z.; Lee, W.X.; Choy, S.M.; Fong, J.X.; Wong, Y.K.; Li, D.; Hu, F.; Tang, H.-W. A Novel TP53 Gene Mutation Sustains Non-Small Cell Lung Cancer through Mitophagy. Cells 2022, 11, 3587.
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141.
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793.
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035.
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269.
- Oeing, C.U.; Nakamura, T.; Pan, S.; Mishra, S.; Dunkerly-Eyring, B.L.; Kokkonen-Simon, K.M.; Lin, B.L.; Chen, A.; Zhu, G.; Bedja, D.; et al. PKG1alpha Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy. Circ. Res. 2020, 127, 522–533.
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244.
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416.
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395.
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597.
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750.
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995.
- Ma, X.; Zhang, S.; He, L.; Rong, Y.; Brier, L.W.; Sun, Q.; Liu, R.; Fan, W.; Chen, S.; Yue, Z.; et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy 2017, 13, 592–607.
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522.
- Koyama-Honda, I.; Itakura, E.; Fujiwara, T.K.; Mizushima, N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013, 9, 1491–1499.
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252.
- Li, L.; Tong, M.; Fu, Y.; Chen, F.; Zhang, S.; Chen, H.; Ma, X.; Li, D.; Liu, X.; Zhong, Q. Lipids and membrane-associated proteins in autophagy. Protein Cell 2021, 12, 520–544.
- Tang, H.W.; Chen, G.C. Unraveling the role of myosin in forming autophagosomes. Autophagy 2011, 7, 778–779.
- Wan, W.; You, Z.; Zhou, L.; Xu, Y.; Peng, C.; Zhou, T.; Yi, C.; Shi, Y.; Liu, W. mTORC1-Regulated and HUWE1-Mediated WIPI2 Degradation Controls Autophagy Flux. Mol. Cell 2018, 72, 303–315.e6.
- Wan, W.; You, Z.; Xu, Y.; Zhou, L.; Guan, Z.; Peng, C.; Wong, C.C.L.; Su, H.; Zhou, T.; Xia, H.; et al. mTORC1 Phosphorylates Acetyltransferase p300 to Regulate Autophagy and Lipogenesis. Mol. Cell 2017, 68, 323–335.e6.
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566.
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787.
- Ding, X.; Jiang, X.; Tian, R.; Zhao, P.; Li, L.; Wang, X.; Chen, S.; Zhu, Y.; Mei, M.; Bao, S.; et al. RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy 2019, 15, 1774–1786.
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337.
- Cheng, X.; Ma, X.; Ding, X.; Li, L.; Jiang, X.; Shen, Z.; Chen, S.; Liu, W.; Gong, W.; Sun, Q. Pacer Mediates the Function of Class III PI3K and HOPS Complexes in Autophagosome Maturation by Engaging Stx17. Mol. Cell 2017, 65, 1029–1043.e5.
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001.
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218.
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290.
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer Is a Mediator of mTORC1 and GSK3-TIP60 Signaling in Regulation of Autophagosome Maturation and Lipid Metabolism. Mol. Cell 2019, 73, 788–802.e7.
- Chang, J.W.; Zhang, W.; Yeh, H.S.; de Jong, E.P.; Jun, S.; Kim, K.H.; Bae, S.S.; Beckman, K.; Hwang, T.H.; Kim, K.S.; et al. mRNA 3’-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015, 6, 7218.
- Tang, H.W.; Hu, Y.; Chen, C.L.; Xia, B.; Zirin, J.; Yuan, M.; Asara, J.M.; Rabinow, L.; Perrimon, N. The TORC1-Regulated CPA Complex Rewires an RNA Processing Network to Drive Autophagy and Metabolic Reprogramming. Cell Metab. 2018, 27, 1040–1054.e8.
- Hu, G.; McQuiston, T.; Bernard, A.; Park, Y.D.; Qiu, J.; Vural, A.; Zhang, N.; Waterman, S.R.; Blewett, N.H.; Myers, T.G.; et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat. Cell Biol. 2015, 17, 930–942.
- Cho, S.; Lee, G.; Pickering, B.F.; Jang, C.; Park, J.H.; He, L.; Mathur, L.; Kim, S.S.; Jung, S.; Tang, H.W.; et al. mTORC1 promotes cell growth via m(6)A-dependent mRNA degradation. Mol. Cell 2021, 81, 2064–2075.e8.
- Tang, H.W.; Weng, J.H.; Lee, W.X.; Hu, Y.; Gu, L.; Cho, S.; Lee, G.; Binari, R.; Li, C.; Cheng, M.E.; et al. mTORC1-chaperonin CCT signaling regulates m(6)A RNA methylation to suppress autophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2021945118.
- Baskin, K.K.; Winders, B.R.; Olson, E.N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 2015, 21, 237–248.
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330.
- Risson, V.; Mazelin, L.; Roceri, M.; Sanchez, H.; Moncollin, V.; Corneloup, C.; Richard-Bulteau, H.; Vignaud, A.; Baas, D.; Defour, A.; et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009, 187, 859–874.
- Bentzinger, C.F.; Romanino, K.; Cloetta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008, 8, 411–424.
- Baraldo, M.; Zorzato, S.; Dondjang, A.H.T.; Geremia, A.; Nogara, L.; Dumitras, A.G.; Canato, M.; Marcucci, L.; Nolte, H.; Blaauw, B. Inducible deletion of raptor and mTOR from adult skeletal muscle impairs muscle contractility and relaxation. J. Physiol. 2022, 600, 5055–5075.
- Pallafacchina, G.; Calabria, E.; Serrano, A.L.; Kalhovde, J.M.; Schiaffino, S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Natl. Acad. Sci. USA 2002, 99, 9213–9218.
- Aguilar, V.; Alliouachene, S.; Sotiropoulos, A.; Sobering, A.; Athea, Y.; Djouadi, F.; Miraux, S.; Thiaudiere, E.; Foretz, M.; Viollet, B.; et al. S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab. 2007, 5, 476–487.
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019.
- Castets, P.; Lin, S.; Rion, N.; Di Fulvio, S.; Romanino, K.; Guridi, M.; Frank, S.; Tintignac, L.A.; Sinnreich, M.; Ruegg, M.A. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013, 17, 731–744.
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.D.; Ji, X.; et al. mTORC1 underlies age-related muscle fiber damage and loss by inducing oxidative stress and catabolism. Aging Cell 2019, 18, e12943.
- Lin, P.W.; Chu, M.L.; Liu, H.S. Autophagy and metabolism. Kaohsiung J. Med. Sci. 2021, 37, 12–19.
- Chen, S.F.; Kang, M.L.; Chen, Y.C.; Tang, H.W.; Huang, C.W.; Li, W.H.; Lin, C.P.; Wang, C.Y.; Wang, P.Y.; Chen, G.C.; et al. Autophagy-related gene 7 is downstream of heat shock protein 27 in the regulation of eye morphology, polyglutamine toxicity, and lifespan in Drosophila. J. Biomed. Sci. 2012, 19, 52.
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515.
This entry is offline, you can click here to edit this entry!