Antimetabolites of folic acid represent a large group of drugs and drug candidates, including those for cancer chemotherapy. Antimetabolites, which are antagonists of natural metabolites, belong to a group of highly efficient anticancer drugs. Based on the chemical structure, these groups can be divided into several sub-groups, such as non-natural amino-acids or peptides, including phospha-analogues, analogues of purine and pyrimidine bases, such as competitors in the synthesis of the nucleic acids, as well as vitamin actions including folic acid, hormones, coenzymes, and other substrates responsible for the normal functioning of cells and tissues of the human body.
- antifolates
- folic acids
- synthesis
1. Introduction
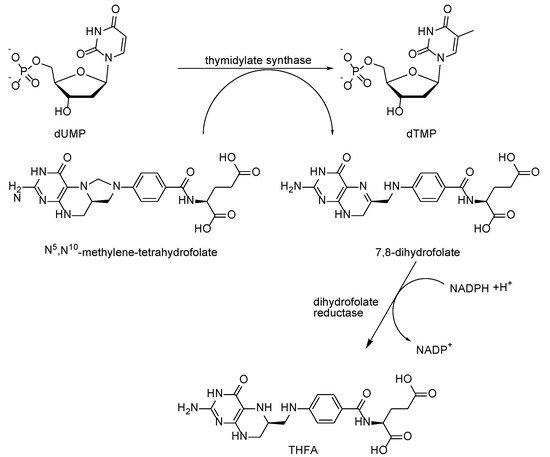
2. Mechanism of Antifolates Action
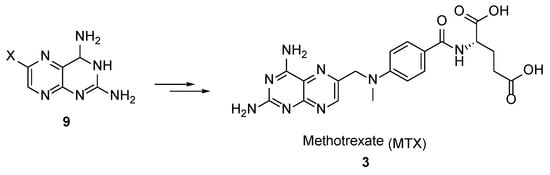
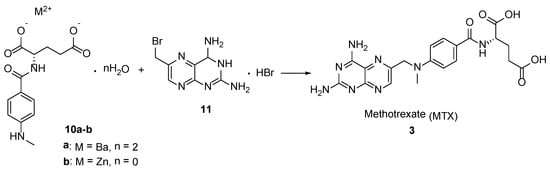
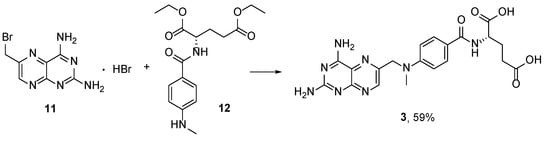
3. Methotrexate: (S)-2-(4-(((2,4-Diaminopteridin-6-yl)Methyl)(Methyl)Amino)benzamido) Pentanedioic Acid (MTX, Rheumatrex, Amethopterin, Abitrexate, Trexall, Methylaminopterin, Mexate, Metatrexan)
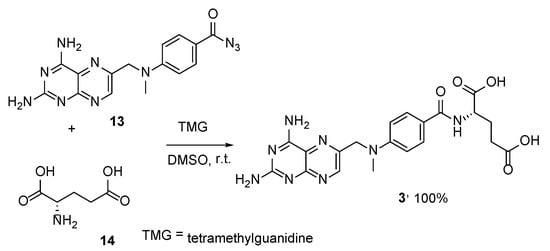
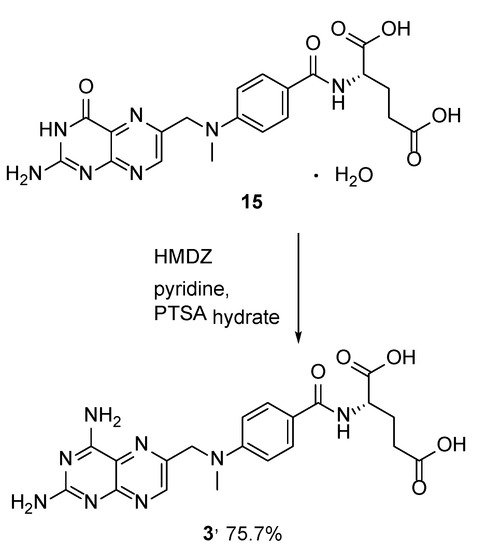
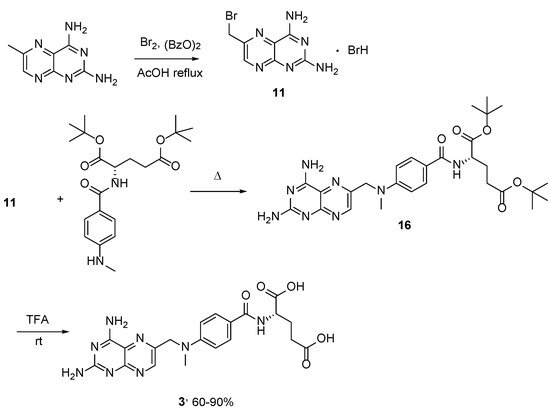
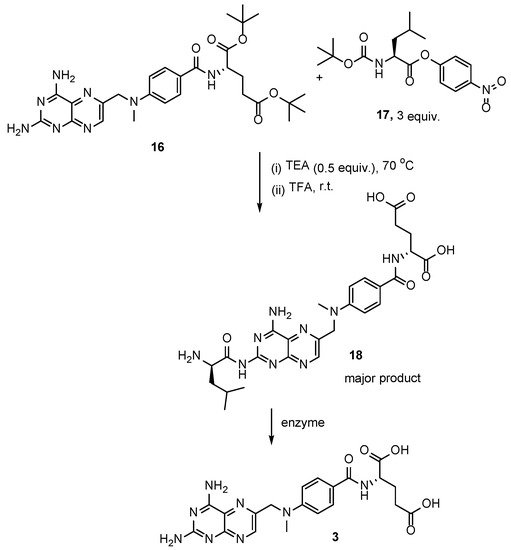
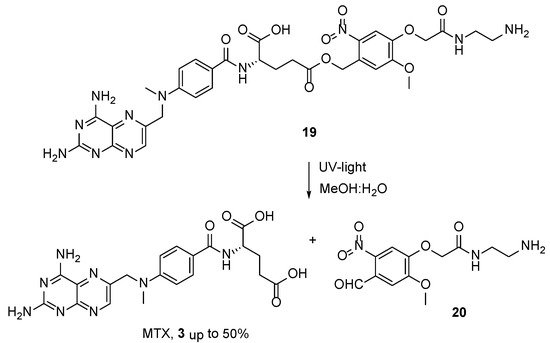
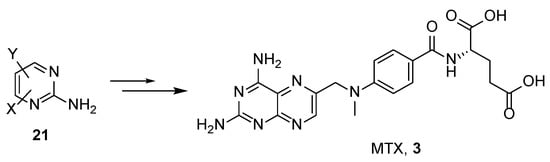
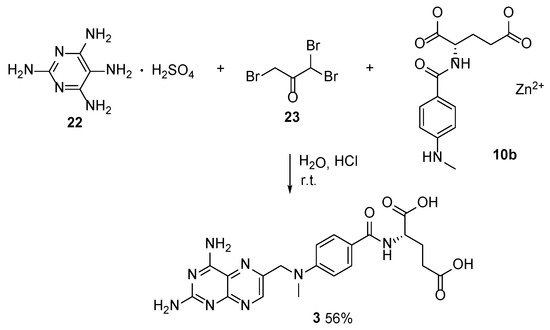
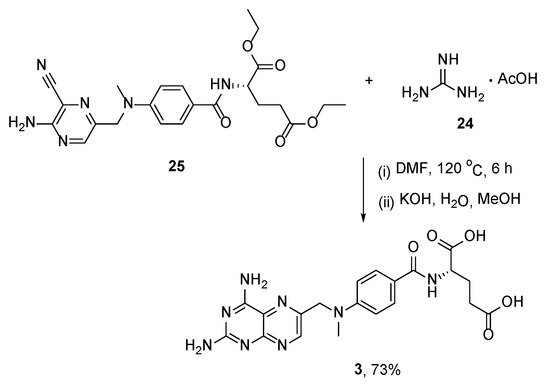
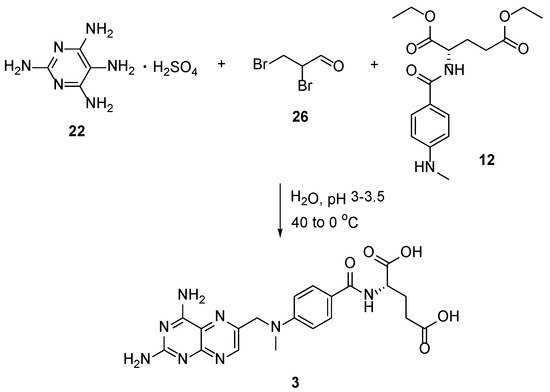
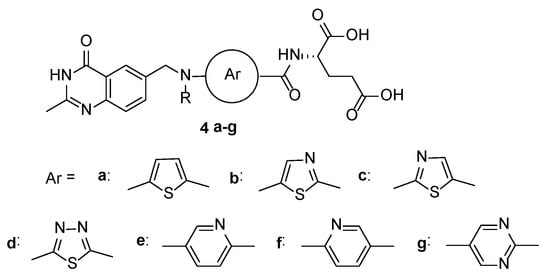
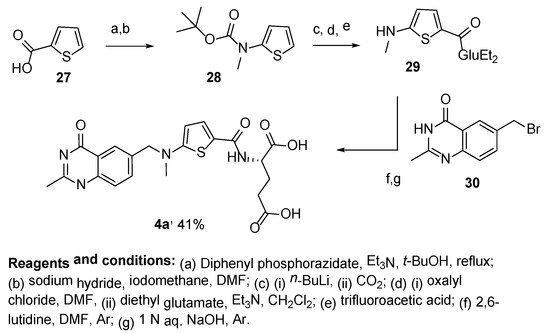
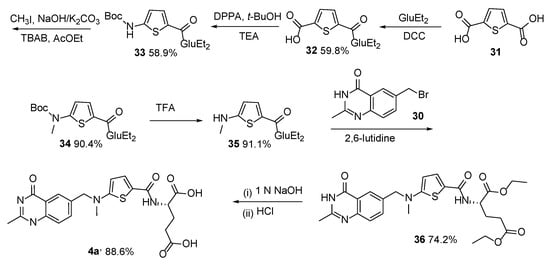
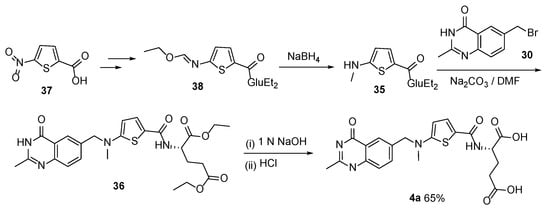
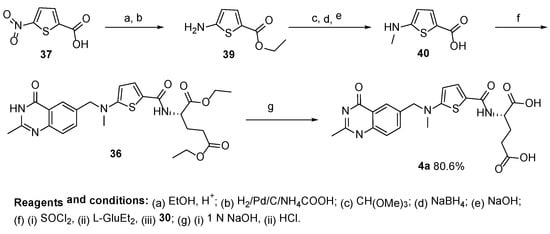
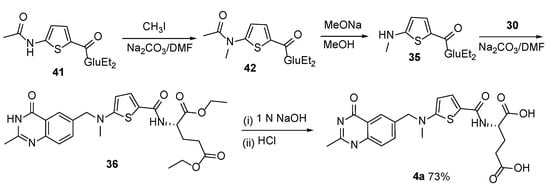
4. Raltitrexed: (2S)-2-[[5-[Methyl-[(2-Methyl-4-oxo-3H-Quinazolin-6-yl)Methyl]Amino] Thiophene-2-Carbonyl]Amino]Pentanedioic Acid (Tomudex, ZD1694)
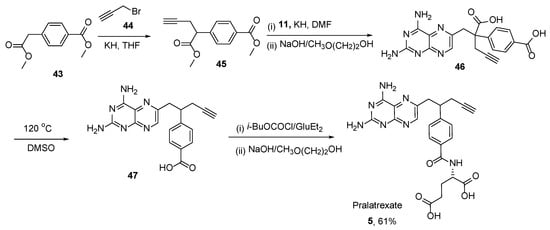
5. Pralatrexate (Folotyn): N-4-[1-(2,4-Diaminopteridin-6-yl)Pent-4-yn-2-yl]Benzoyl-L-Glutamic Acid
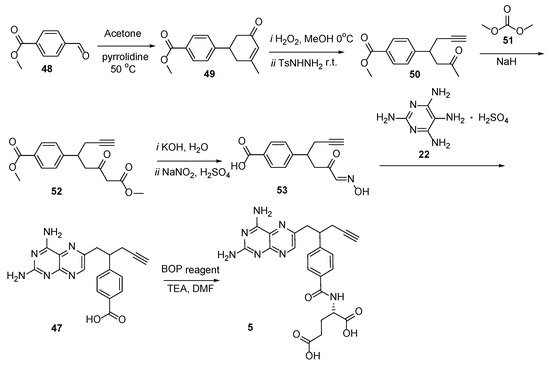
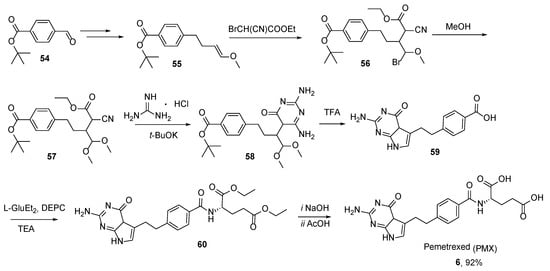
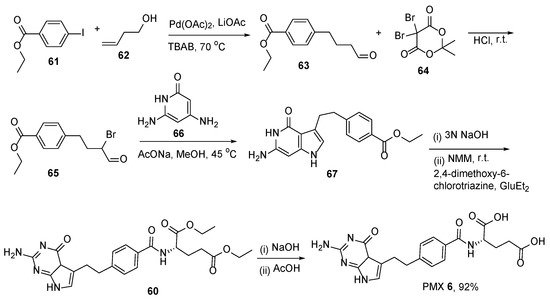
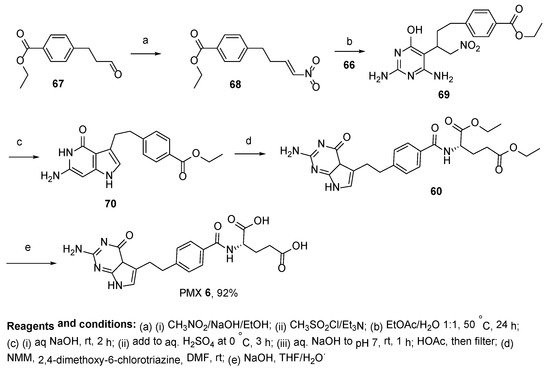
6. Pemetrexed: (S)-2-(4-(2-(2-Amino-4-oxo-4,7-Dihydro-1H-Pyrrolo[2,3-d]Pyrimidin-5-yl)Ethyl)Benzamido)Pentanedioic Acid (PMX, ALIMTA, LY231514, MTA)
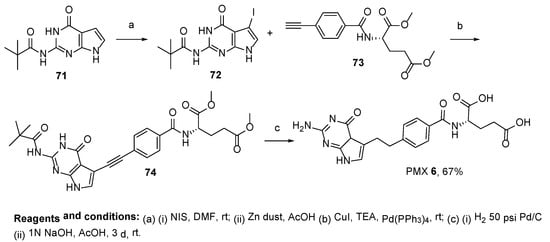
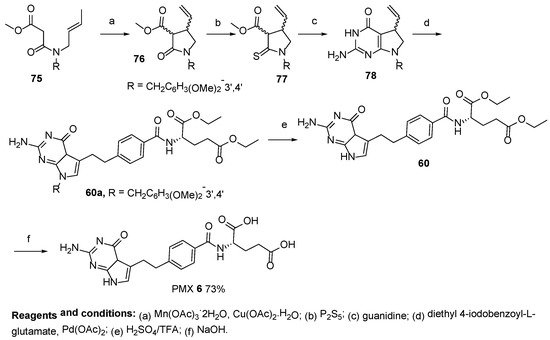
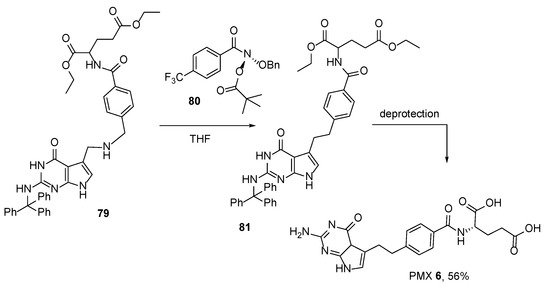
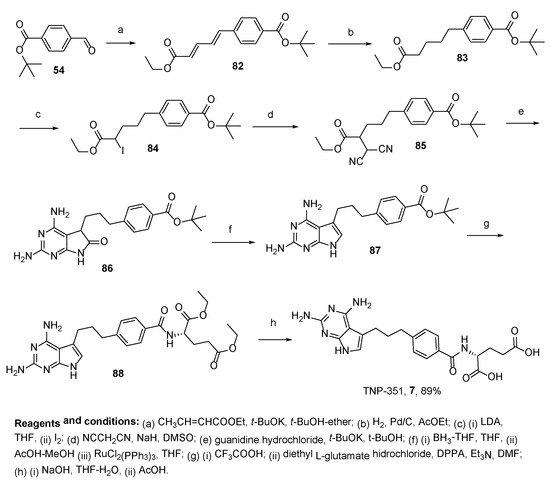
7. TNP-351: (2S)-2-[[4-[3-(2,4-Diamino-7H-Pyrrolo[2,3-d]Pyrimidin-5-yl)Propyl]benzoyl]Amino]Pentanedioic Acid (HY-19095)
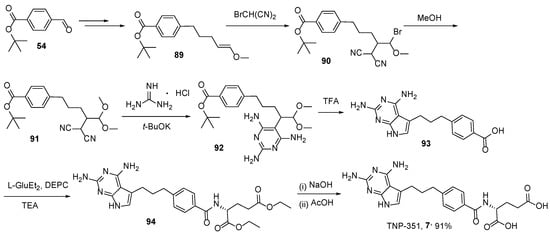
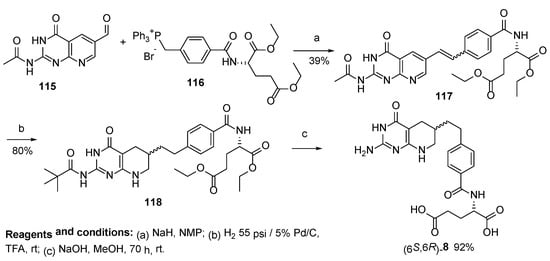
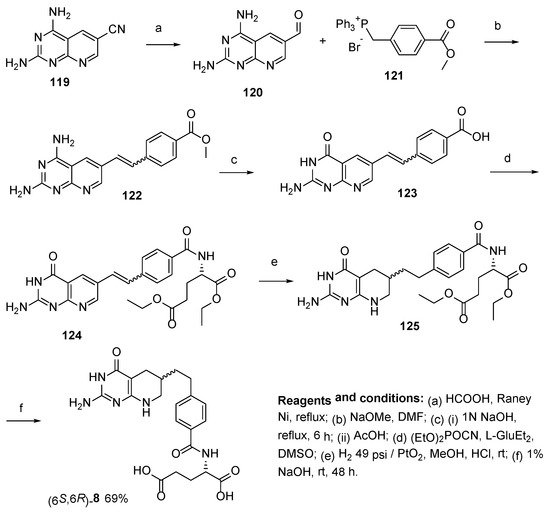
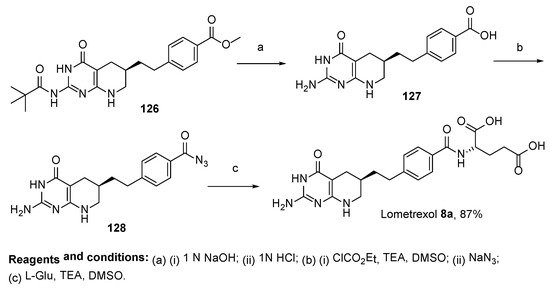
8. Lometrexol: (2S)-2-[[4-[2-[(6R)-2-Amino-4-oxo-5,6,7,8-Tetrahydro-1H-Pyrido[2,3-d]Pyrimidin-6-yl]Ethyl]Benzoyl]Amino]Pentanedioic Acid (LY 264618, DDATHF-B, Lometrexolum)
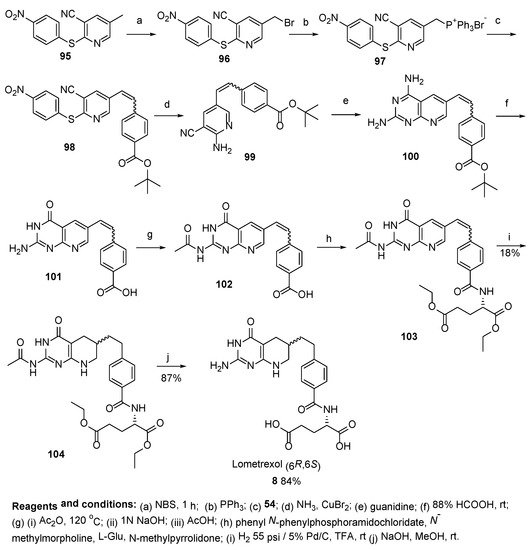
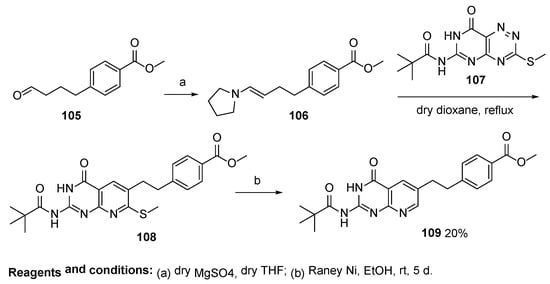
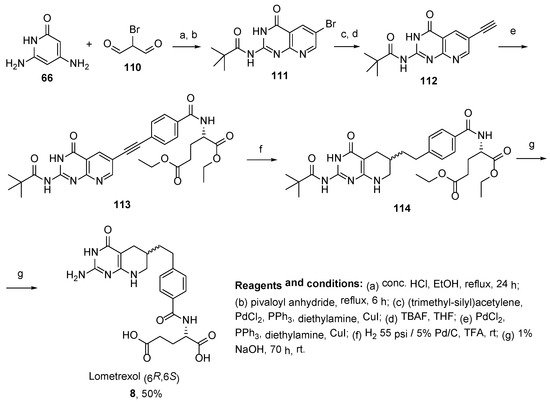
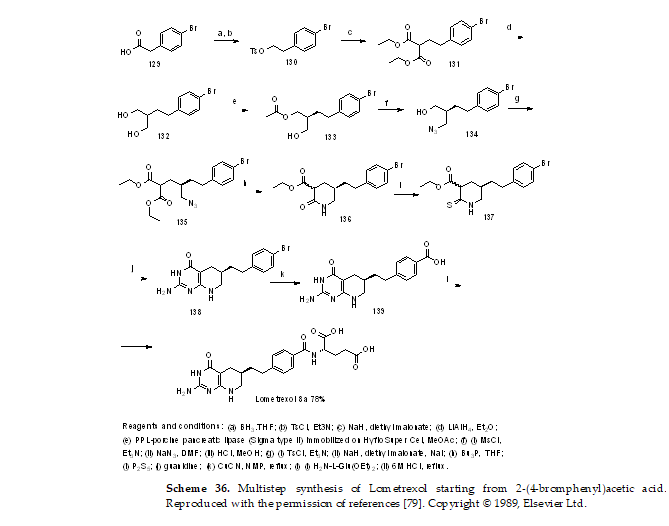
Scheme 36. Multistep synthesis of Lometrexol starting from 2-(4-bromphenyl)acetic acid. Reproduced with the permission of references. Copyright © 1989, Elsevier Ltd.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27196229
References
- Ahluwalia, G.S.; Grem, J.L.; Hao, Z.; Cooney, D.A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271.
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004.
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application (Review). Int. J. Oncol. 2020, 57, 678–696.
- Mandal, P.K.; Gao, F.; Lu, Z.; Ren, Z.; Ramesh, R.; Birtwistle, J.S.; Kaluarachchi, K.K.; Chen, X.; Bast, R.C., Jr.; Liao, W.S.; et al. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J. Med. Chem. 2011, 54, 3549–3563.
- Gut, J. Aza Analogs of Pyrimidine and Purine Bases of Nucleic Acids. Adv. Heterocycl. Chem. 1963, 1, 189–251.
- Gojkovic, Z.; Karlsson, A. Purine and Pyrimidine-Based Analogs and Suicide Gene Therapy. In Deoxynucleoside Analogs in Cancer Therapy; Humana Press: Totowa, NJ, USA, 2007; pp. 403–439.
- Zelder, F.; Sonnay, M.; Prieto, L. Antivitamins for medicinal applications. ChemBioChem 2015, 16, 1264–1278.
- Howell, A. The Role of Antihormones; CRC Press: Boca Raton, FL, USA, 1990.
- Albert, A. Antimetabolites: Antagonistic analogues of coenzymes and enzyme substrates. In Selective Toxicity; Albert, A., Ed.; Springer: Dordrecht, The Netherlands, 1985; pp. 323–378.
- Lansiaux, A. Les antimétabolites. Bull. Cancer 2011, 98, 1263–1274.
- Chu, E.; Drake, J.C.; Boarman, D.; Baram, J.; Allegra, C.J. Mechanism of thymidylate synthase inhibition by methotrexate in human neoplastic cell lines and normal human myeloid progenitor cells. J. Biol. Chem. 1990, 265, 8470–8478.
- Messmann, R.; Allegra, C. Antifolates. In Cancer Chemotherapy and Biotherapy; Chabner, B., Longo, D., Eds.; L. Williams & W: Philadelphia, PA, USA, 2001; pp. 139–184.
- Shih, C.; Chen, V.J.; Gossett, L.S.; Gates, S.B.; MacKellar, W.C.; Habeck, L.L.; Shackelford, K.A.; Mendelsohn, L.G.; Soose, D.J.; Patel, V.F.; et al. Ly231514, a pyrrolo pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997, 57, 1116–1123.
- Avendaño, C.; Menéndez, J.C. (Eds.) Antimetabolites. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008; pp. 9–52.
- Moran, R.G. Folate antimetabolites inhibitory to de novo purine synthesis. In New Drugs, Concepts and Results in Cancer Chemotherapy. Cancer Treatment and Research; Muggia, F.M., Ed.; Springer: Boston, MA, USA, 1991; Volume 58, pp. 65–87.
- Catalucci, E. Process for the Production of Methotrexate. U.S. Patent US4224446A, 23 September 1991.
- Bin, Y.; Shubin, W.; Zhichao, M.; Quansheng, S.; Yanjiao, X.; Lilian, L. A Kind of Preparation Method of Methotrexate (MTX). U.S. Patent CN109553619A, 2 April 2019.
- Piper, J.R.; Montgomery, J.A. 6-(Bromomethyl)-2,4-diaminopteridine Hydrobromide. U.S. Patent US4077957A, 11 April 1978.
- Luo, J.; Smith, M.D.; Lantrip, D.A.; Wang, S.; Fuchs, P.L. Efficient syntheses of pyrofolic acid and pteroyl azide, reagents for the production of carboxyl-differentiated derivatives of folic acid. J. Am. Chem. Soc. 1997, 119, 10004–10013.
- Attoline, E.; Michieletti, M.; Rossi, D.; Allegrini, P. Process for the Preparation of Pteridine Derivatives. U.S. Patent US4767859, 17 November 1988.
- Cheung, A.H.T.; Smal, M.; Chau, D.D. Synthesis of multi-13C-enriched methotrexate for NMR studies of drug-Enzyme interactions. Heterocycles 1987, 25, 507–514.
- Cheung, A.H.T.; Boadle, D.K.; Tran, T.Q. N-(L-α-aminoacyl) derivatives of methotrexate. Heterocycles 1989, 28, 751–758.
- Choi, S.K.; Verma, M.; Silpe, J.; Moody, R.E.; Tang, K.; Hanson, J.J.; Baker, J.R., Jr. A photochemical approach for controlled drug release in targeted drug delivery. Bioorg. Med. Chem. 2012, 20, 1281–1290.
- Lei, T.; Yi, Z.; Jianta, W.; Gaofeng, Z.; Xing, C. Synthesis Process of Methotrexate. U.S. Patent CN112851676A, 28 May 2021.
- Wiecko, J. Process for Preparing Methotrexate or an N-Substituted Derivative Thereof and/or a di (lower) Alkyl Ester Thereof and Precursor Therefor. U.S. Patent US4057548A, 30 March 1976.
- Seeger, D.R.; Cosulich, D.B.; Smith, J.M.; Hultquist, M.E. Analogs of Pteroylglutamic Acid. III. 4-Amino Derivatives. J. Am. Chem. Soc. 1949, 71, 1753–1758.
- Widemann, B.C.; Balis, F.M.; Godwin, K.S.; McCully, C.; Adamson, P.C. The plasma pharmacokinetics and cerebrospinal fluid penetration of the thymidylate synthase inhibitor raltitrexed (Tomudex(TM)) in a nonhuman primate model. Cancer Chemother. Pharmacol. 1999, 44, 439–443.
- Liu, Y.; Wu, W.; Hong, W.; Sun, X.; Wu, J.; Huang, Q. Raltitrexed-based chemotherapy for advanced colorectal cancer. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 219–225.
- Cocconi, G.; Cunningham, D.; Van Cutsem, E.; Francois, E.; Gustavsson, B.; van HazelD Kerr, G.; Possinger, K.; Hietschold, S.M. Open, randomized, multicenter trial of raltitrexed versus fluorouracil plus high-dose leucovorin in patients with advanced colorectal cancer. Tomudex Colorectal Cancer Study Group. J. Clin. Oncol. 2016, 16, 2943–2952.
- Marsham, P.R.; Hughes, L.R.; Jackman, A.L.; Hayter, A.J.; Oldfield, J.; Wardleworth, J.M.; Bishop, J.A.M.; O’Connor, B.M.; Calvert, A.H. Quinazoline Antifolate Thymidylate Synthase Inhibitors: Heterocyclic Benzoyl Ring Modifications. J. Med. Chem. 1991, 34, 1594–1605.
- Cao, S.L.; Wan, R.; Feng, Y.P. New synthesis of thymidylate synthase inhibitor raltitrexed. Synth. Commun. 2003, 33, 3519–3526.
- Yao, T.; Zubing, W.; Shuwang, G.; Jian, W.; Yuzhu, C.; Feng, L.; Dan, X.; Chunxia, Z.; Zhoushan, T. The Formoxyl of 5-((Alkoxy methylene)amino)thienyl-2-formyl Group)-L-glutamic Acid Dialkyl Ester and Preparation Method Thereof. U.S. Patent CN106957296A, 18 July 2017.
- Xiqun, J.; Wei, W.; Zhoushan, T.; Jun, H.; Jing, W.; Yuzhu, C.; Huaping, W.; Dan, X.; Chunxia, Z.; Chuanjun, W. A kind of Pharmaceutical Composition of Raltitrexed Pharmaceutical Composition and Preparation Method Thereof. U.S. Patent CN107616976A, 23 January 2018.
- Shaojie, H.; Wei, S.; Zhaobai, Z.; Xu, S. Preparation Method of Raltitrexed. U.S. Patent CN110551114A, 10 December 2019.
- O’Connor, O.A.; Pro, B.; Pinter-Brown, L.; Bartlett, N.; Popplewell, L.; Coiffier, B.; Lechowicz, M.J.; Savage, K.J.; Shustov, A.R.; Gisselbrecht, C.; et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: Results from the pivotal PROPEL study. J. Clin. Oncol. 2011, 29, 1182–1189.
- Amengual, J.E.; Lichtenstein, R.; Lue, J.; Sawas, A.; Deng, C.; Lichtenstein, E.; Khan, K.; Atkins, L.; Rada, A.; Kim, H.A.; et al. A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 2018, 131, 397–407.
- Parker, T.; Barbarotta, L.; Foss, F. Pralatrexate: Treatment of T-cell non-Hodgkin’s lymphoma. Future Oncol. 2012, 9, 21–29.
- DeGraw, J.I.; Colwell, W.T.; Piper, J.R.; Sirotnak, F.M. Synthesis and Antitumor Activity of 10-Propargyl-10-deazaaminopterin. J. Med. Chem. 1993, 36, 2228–2231.
- Guimin, Z.; Chengfu, C.; Chuanbing, W. A Kind of Preparation Method of Pralatrexate. U.S. Patent CN108069970, 02 June 2020.
- Giust, W.; Burton, R.; Gorin, B.; Clayton, J. Process for Preparation of an Antifolate Agent. U.S. Patent WO2013/177713A1, 5 December 2013.
- O’Connor, O.A.; Sirotnak, F.M. Treatment of T-Cell Lymphoma Using 10-Propargyl-10-Deazaaminopterin. U.S. Patent US2005/267117A1, 1 December 2005.
- Lahiri, S.; Gupta, N.; Singh, H.K.; Panda, N.; Handa, V.; Abul, A.; Gupta, C.K.; Sanghani, S.; Sonavane, G.M. Process for the Preparation of Pralatrexate. U.S. Patent US9440979B2, 13 September 2016.
- Lahiri, S.; Gupta, N.; Singh, H.K. Salts of Pralatrexate. U.S. Patent WO2014/20553A1, 6 February 2014.
- Lahiri, S.; Gupta, N.; Singh, H.K.; Panda, N.; Handa, V.; Abul, A.; Gupta, C.K.; Sanghani, S.; Sonavane, G.M. Improved Process for the Preparation of Pralatrexate. U.S. Patent WO2014016740A2, 30 January 2014.
- Tiseni, P.S.; Galluzzo, C.; Canavesi, A.; Biljan, T. Processes and Intermediates for Preparing Pralatrexate. U.S. Patent EP2794610B1, 27 June 2013.
- Pronk, G.J. Optically Pure Diastereomers of 10-Propargyl-10-Deazaaminopterin and Methods of Using Same. U.S. Patent US8835433B2, 4 August 2011.
- Alla, R.R.V.; Ramarao, C.; Michel, P.T.; Nitlikar, L.H.; Kalam, B.R.; Duduka, R. A Process for Preparing Intermediates of 10-propargyl-10-deazaaminopterin (pralatrexate) Synthesis and the Intermediates Thereof. U.S. Patent WO2013164856A1, 7 November 2013.
- Cohen, M.H.; Justice, R.; Pazdur, R. Approval Summary: Pemetrexed in the Initial Treatment of Advanced/Metastatic Non-Small Cell Lung Cancer. Oncologist 2009, 14, 930–935.
- Rossi, A.; Ricciardi, S.; Maione, P.; de Marinis, F.; Gridelli, C. Pemetrexed in the treatment of advanced non-squamous lung cancer. Lung Cancer 2009, 66, 141–149.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Azzoli, C.G.; Kris, M.G.; Pfister, D.G. Cisplatin versus carboplatin for patients with metastatic non-small-cell lung cancer–an old rivalry renewed. J. Natl. Cancer Inst. 2007, 99, 828–829.
- Manegold, C. Pemetrexed (alimta, MTA, multitargeted antifolate, LY231514) for malignant pleural mesothelioma. Semin. Oncol. 2003, 30, 32–36.
- McLeod, H.L.; Cassidy, J.; Powrie, R.H.; Priest, D.G.; Zorbas, M.A.; Synold, T.W.; Shibata, S.; Spicer, D.; Bissett, D.; Pithavala, Y.K.; et al. Pharmacokinetic and Pharmacodynamic Evaluation of the Glycinamide Ribonucleotide Formyltransferase Inhibitor AG20341. Clin. Cancer Res. 2000, 6, 2677–2684.
- Miwa, T.; Hitaka, T.; Akimoto, H. A Novel Synthetic Approach to Pyrrolo pyrimidine Antifolates. J. Org. Chem. 1993, 58, 1696–1701.
- Mitchell-Ryan, S.; Wang, Y.; Raghavan, S.; Ravindra, M.P.; Hales, E.; Orr, S.; Cherian, C.; Hou, Z.; Matherly, L.H.; Gangjee, A. Discovery of 5-substituted pyrrolopyrimidine antifolates as dual-acting inhibitors of glycinamide ribonucleotide formyltransferase and 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase in de novo purine nucleotide biosynthesis. J. Med. Chem. 2013, 56, 10016–10032.
- Bonaccorsi, F.; Calvani, F.; Pasqui, F. Process for the Preparation of Pemetrexed and Lysin Salt Thereof. U.S. Patent EP2882753B1, 13 February 2014.
- Michalak, O.; Gruza, M.M.; Witkowska, A.; Bujak, I.; Cmoch, P. Synthesis and physicochemical characterization of the impurities of pemetrexed disodium, an anticancer drug. Molecules 2015, 20, 10004–10031.
- Taylor, E.C.; Liu, B. A simple and concise synthesis of LY231514 (MTA). Tetrahedron Lett. 1999, 40, 4023–4026.
- Taylor, E.C.; Liu, B. A New and Efficient Synthesis of Pyrrolopyrimidine Anticancer Agents: Alimta (LY231514, MTA), Homo-Alimta, TNP-351, and Some Aryl 5-Substituted Pyrrolopyrimidines. J. Org. Chem. 2003, 68, 9938–9947.
- Taylor, E.C.; Liu, B. Process for the Preparation of pyrrolopyrimidines. U.S. Patent US6066732 A, 23 May 2000.
- Taylor, E.C.; Kuhnt, D.; Shih, C.; Rinzel, S.M.; Grindey, G.B.; Barredo, J.; Jannatipour, M.; Moran, R.G. A Dideazatetrahydrofolate Analogue Lacking a Chiral Center at C-6, N-pyrimidin-5yl)ethyl-l-glutamic Acid is an Inhibitor of Thymidylate Synthase. J. Med. Chem. 1992, 35, 4450–4454.
- Taylor, E.C.; Liu, B. A novel synthetic route to 7-substituted derivatives of the antitumor agent LY231514 (MTA). Tetrahedron Lett. 1999, 40, 5291–5294.
- Kennedy, S.H.; Dherange, B.D.; Berger, K.J.; Levin, M.D. Skeletal editing through direct nitrogen deletion of secondary amines. Nature 2021, 593, 223–227.
- Itoh, F.; Russello, O.; Akimoto, H.; Beardsley, G.P. Novel pyrrolopyrimidine antifolate TNP-351: Cytotoxic effect on methotrexate-resistant CCRF-CEM cells and inhibition of transformylases of de novo purine biosynthesis. Cancer Chemother. Pharmacol. 1994, 34, 273–279.
- Miwa, T.; Hitaka, T.; Akimoto, H.; Nomura, H.J. Novel pyrrolo pyrimidine antifolates: Synthesis and antitumor activities. Med. Chem. 1991, 34, 555–560.
- Adams, J.; Elliott, P.J. New agents in cancer clinical trials. Oncogene 2000, 19, 6687–6692.
- Newell, D.R. Clinical pharmacokinetics of antitumor antifolates. Semin. Oncol. 1999, 26, 74–81.
- Bronder, J.L.; Moran, R.G. Antifolates targeting purine synthesis allow entry of tumor cells into S phase regardless of p53 function. Cancer Res. 2002, 62, 5236–5241.
- Taylor, E.C.; Harrington, P.J.; Fletcher, S.R.; Beardsley, G.P.; Moran, R.G. Synthesis of the Antileukemic Agents 5, 10-Dideazaaminopterin and 5, 10-Dideaza-5, 6, 7, 8-tetrahydroaminopterin. J. Med. Chem. 1985, 28, 914–921.
- Taylor, E.C.; Harrington, P.M.; Warner, J.C. Diels-Alder reactions of 6-azapterins. An alternative strategy for the synthesis of 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Heterocycles 1988, 27, 1925–1928.
- Taylor, E.C.; Wong, G.S.K. Convergent and Efficient Palladium-Effected Synthesis of 5,10-Dideaza-5,6,7,8-tetrahydrofolic Acid (DDATHF). J. Org. Chem. 1989, 54, 3618–3624.
- Boschelli, D.H.; Webber, S.; Whiteley, J.M.; Oronsky, A.L.; Kerwar, S.S. Synthesis and biological properties of 5,10-dideaza-5,6,7,8-tetrahydrofolic acid. Arch. Biochem. Biophys. 1988, 265, 43–49.
- Piper, J.R.; McCaleb, G.S.; Montgomery, J.A.; Kisliuk, R.L.; Gaumont, Y.; Thorndike, J.; Sirotnak, F.M. Synthesis and Antifolate Activity of 5-Methyl-5,10-dideaza Analogues of Aminopterin and Folic Acid and an Alternative Synthesis of 5,10-Dideazatetrahydrofolic Acid, a Potent Inhibitor of Glycinamide Ribonucleotide Formyltransferase. J. Med. Chem. 1988, 31, 2164–2169.
- Taylor, E.C.; Chaudhari, R.; Lee, K. A simplified and efficient synthesis of 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Investig. New Drugs 1996, 14, 281–285.
- Tomsho, J.W.; McGuire, J.J.; Coward, J.K. Synthesis of (6R)- and (6S)-5,10-dideazatetrahydrofolate oligo-γ-glutamates: Kinetics of multiple glutamate ligations catalyzed by folylpoly-γ-glutamate synthetase. Org. Biomol. Chem. 2005, 3, 3388–3398.
- Barnett, C.J.; Wilson, T.M. Asymmetric synthesis and absolute configuration of 5,10-dideaza-5,6,7,8-tetrahydropteroic acid and 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Tetrahedron Lett. 1989, 30, 6291–6294.
- Barnett, C.J.; Wilson, T.M.; Wendel, S.R.; Winningham, M.J.; Deeter, J.B. Asymmetric Synthesis of Lometrexol ((6R)-5,10-Dideaza-5,6,7,8-tetrahydrofolic Acid). J. Org. Chem. 1994, 59, 7038–7045.