Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Tumor cell-derived extracellular vesicles (TEVs) are an important means of tumor communication with, and manipulation of, the patient’s physiology. TEVs influence the local tumor environment as well as the systemic conditions of the patient.
- exosomes
- immunoediting
- cancer immunity
- immune escape
- immunosurveillance
- tumor microenvironment
1. Introduction
The tumor microenvironment (TME) within and around a tumor is a complex interacting mixture of tumor cells with various stromal cells, including endothelial cells, fibroblasts, and immune cells. In the early steps of tumor formation, the local microenvironment tends to oppose carcinogenesis, while with cancer progression, the microenvironment skews into a protumoral TME and the tumor influences stromal cells to provide tumor-supporting functions. The creation and development of cancer are dependent on escape from immune recognition predominantly by influencing stromal cells, particularly immune cells, to suppress antitumor immunity. This overall process is generally called immunoediting and has been categorized into three phases; elimination, equilibrium, and escape. Interaction of tumor cells with stromal cells in the TME is mediated generally by cell-to-cell contact, cytokines, growth factors, and extracellular vesicles (EVs). The least well studied are EVs (especially exosomes), which are nanoparticle-sized bilayer membrane vesicles released by many cell types that participate in cell/cell communication. EVs carry various proteins, nucleic acids, lipids, and small molecules that influence cells that ingest the EVs. Tumor-derived extracellular vesicles (TEVs) play a significant role in every stage of immunoediting, and their cargoes change from immune-activating in the early stages of immunoediting into immunosuppressing in the escape phase.
TEV early in tumor development can stimulate antitumor immunity. The interaction between immune cells and cancer in TME is categorized into seven potential steps (Figure 1) [4] which is called the “cancer-immunity cycle”. In the right conditions, TEV from tumor cells can also support antitumor immunity. TEVs contain and transfer TAs and damage-associated molecular patterns (DAMPs) to innate immune cells, especially dendritic cells (Step 1) [85,86]. Tumor-derived EVs are a source of shared TAs for CTL cross-priming.
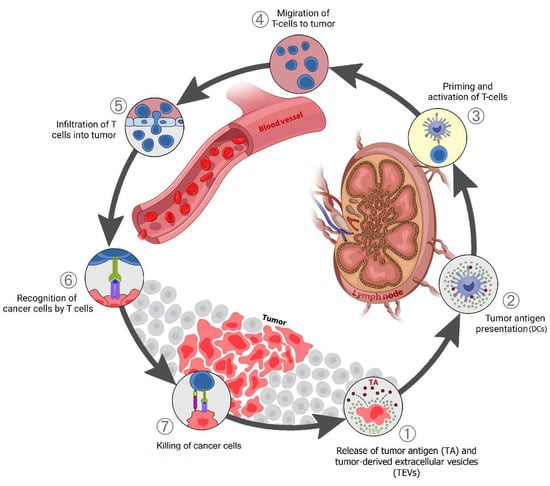
Figure 1. TEV in the cancer-immunity cycle: (1) Release of tumor antigens (TAs) along with tumor-derived extracellular vesicles (TEVs) that carry TA + DAMPs from dying cancer cells; (2) Presentation of TAs on the major histocompatibility complex (MHC) by dendritic cells; (3) T-cell receptor recognition of TAs on the MHC, leading to T-cell activation; (4) Migration of activated T cells to the tumors; (5) T-cell infiltration into the tumor; (6) Recognition of cancer antigens within the tumor; (7) Attack and killing of tumor cells.
Dendritic cells (DCs) respond to TEVs carrying DAMPs and TAs, mature, and migrate to lymph nodes (Steps 1–2). The tumor antigen is cross-presented on MHC class I (MHC-I) in the lymph nodes where it activates naive CD8 T cells (Step 3). The activated effector T cells go to the tumor site (Step 4), penetrate the tumor tissue (Step 5), identify cancer cells by tumor antigens presented on MHC-I (Step 6), then attack and kill them (Step 7). One or more of these stages may be disrupted in many cancer patients, resulting in ineffective immune responses to cancer. Disruption at any stage of this cycle is caused by cancer cells and their secreted factors, including via TEVs [86,87]. This disruption and immune system suppression block antitumor immunity and support cancer progression.
2. Elimination Phase TEV Involvement
This phase has not been directly detected in vivo in humans since it occurs with very small tumors. The innate and adaptive immune systems collaborate to identify and eliminate tumors that have evaded intrinsic tumor suppressor mechanisms in developing tumors [14]. Cancer immunosurveillance is proposed to remove newly generated neoplastic cells that have the potential to develop tumors. The processes of how the immune system is alerted of the presence of primary tumor cells remain unknown. Among the possibilities, the generation of neoantigens by abnormal cells within the created inflammatory environment (via immune cell subsets, recognition molecules, and effector cytokines) results in the detection of nascent cancers, and the traditional warning signals such as IFNs are likely involved [88,89,90]. T cells are the primary immune cells that identify and eliminate tumor cells [88,91]. However, B cells and their antibodies also seem to play a role in recognizing and removing these cells [92]. IFN-γ has a direct anti-proliferative impact on tumors through the STAT1 pathway and causes the release of cytokines such as CXCL9, 10, and 11 that increase immune activation by recruiting effector T cells [93,94]. IFN alpha and beta (type I IFNs) also play an important role in activating CD103+ DCs to cross-present tumor antigens [15,95].
Physical characteristics of the tumor environment such as hypoxia can cause tumor cell death, potentially resulting in the release of DAMPs such as Heat shock proteins (HSPs) and high mobility group box 1 (HMGB1), which act as ligands for Toll-like receptors on innate immune cells [85]. EVs can carry TAs, interferon, and DAMPs that stimulate immunological responses against tumors [96,97]. EVs carried CEA and HER2 TAAs that triggered immune responses and improved anti-tumor responses in vivo [98]. The release of tumor antigens and EVs can be altered under various TME situations. For example, an acidic microenvironment, quite common for tumors, increased the number of secreted EVs [99].
TEVs can play a key role in NK cell activation, DC maturation, and CD8+ effector T-cell development [100,101]. TEVs may also carry surface proteins derived from cancer cells which promote the uptake of TEVs by DCs. There are reports supporting LFA-1/CD54 and mannose-rich C-type lectin receptor interactions as enabling TEV uptake by DCs [102,103]. Uptake of TEVs by DCs enhanced DC expression of co-stimulatory receptors such as CD80, CD86, and also MHC II expression and boosted interferon and cytokine production along with DC maturation [104,105,106]. Breast cancer cells generated EVs that convey dsDNA to DCs, causing IFN alpha and beta expression in a STING-dependent manner and elevation of costimulatory molecules in DCs [107].
Furthermore, TEVs carry molecules that promoted CD8+ T-cell activation and enhanced tumor cytotoxic T lymphocyte (CTL) responses in vivo in mice [105,106,108]. EVs generated from brain tumors were delivered to mice on days 7 and 14 post-tumor inoculation, stimulating antibody production and T-cell activation. Antitumor antibodies and T cells present at the time of tumor inoculation appear to have caused enough tumor cell death to generate further T-cell antitumor response [109].
Tumor-Derived Immunostimulatory Vesicular DAMPs
During an immunogenic cell death (ICD), cancer cells release danger signals (DAMPs) raising the immunogenicity of dying cancer cells [85,110,111,112]. ICD is more immune stimulatory than necrosis which can suppress immunological responses [113] and necrosis generally does not strongly stimulate CD8+ T-cell-dependent immune responses [114]. DAMPs are secreted as a result of endoplasmic reticulum (ER) stress induced by mitochondrial ROS, membrane-lipid peroxidation, and ER-directed ROS generation [115,116,117]. DAMPs can also be released during necroptosis, pyroptosis, and ferroptosis [118,119,120]. EVs from cancer cells can carry DAMPs including HSPs, HMGB1, histones, ATP, vesicular RNAs, and cell-free DNA inside or on the surface [121,122,123,124,125,126]. Interestingly, EVs with surface-bound HSP70 stimulate more helper T cells (Th1) and CTL than TEVs with cytoplasmic HSP70 inside EVs [126]. Hsp70-enriched TEVs elicited significant CD4+ Th1 immune responses and promoted the production of MHC class II molecules on antigen-presenting cells, leading to the elimination of cancer cells [127]. CD94+ NK cells in the presence of TEVs possessing membrane HSP70 released granzyme B [126,128] and expressed stimulating receptors such as the NKG2D, CD69, and NKp44 while also down-regulating inhibitory receptor CD94 [129].
3. Equilibrium Phase TEV Involvement
Molecular processes that initiate immune-mediated cancer dormancy/control, i.e., the equilibrium phase (EqP), are not well understood in part because this phase is hard to model and has been minimally characterized in humans [130]. Not surprisingly, when overall mechanisms are poorly understood, there is not much known about the involvement of EVs in the equilibrium phase. In the equilibrium phase, the adaptive effector functions and the resistance of the tumor are in a dynamic balance. There are clear indications that tumors in the escape phase having metastasized, can return to equilibrium following chemotherapy and be dormant for many years before relapse. This occurs in particular with metastatic breast tumors where metastatic cells stop proliferating but survive in a quiescent state [131]. The role, if any that the immune system plays in maintaining this dormancy is not clear.
In the EqP, TEVs may suppress different adaptive immune cell types through various mechanisms such as inhibiting effector cells such as CD8+ T cells and NK cells, suppressing DC maturation and activation, increasing M2 and TAM immune suppressive polarization, and stimulating CAF differentiation [64,132,133]. However, as noted previously, TEV can also mediate tumor-suppressing signals. TEVs containing miR-23b derived from mesenchymal bone marrow cancer stem cells (CSC) can induce cancer dormancy via downregulation of the MARCKS gene that mediates breast cancer cells’ differentiation into CSCs through the Wnt-β-catenin pathway [134,135].
Considering PD-L1 and IFN-γ in the EqP of tumors is of interest for understanding the involvement of TEV and highlighting the complexity of molecular interactions. While IFN-γ supports CD8 T-cell effector function, IFN-γ stimulation also increases the quantity of PD-L1 on melanoma-released EVs that in turn suppressed the effector function of CD8+ T cells [136]. IFN-γ induced tumor dormancy when the interferon-gamma receptor 1 (IFNGR1) expression level was low but resulted in tumor elimination when it was high [137]. GW4869 treatment or Rab27a knockdown can inhibit vesicular-PD-L1 secretion, and significantly augment anti-PD-L1 therapeutic efficacy in 4T1 tumor growth [138]. Animal studies have shown that TEVs can also impair the production of interferons as well as decrease innate immune activity via EGFR- and MEKK2- dependent pathways [139].
4. Escape Phase TEV Involvement
Clinically recognized tumors have generally moved from equilibrium to escape. In the equilibrium phase, genome instability and accumulation of mutations in cancer cells over time leads to selection for low immunogenicity, expression of immune suppressive ligands, and escape from the immune system [140]. Tumors can eventually overcome antitumor immunity through mechanisms already mentioned, including tumor antigen editing, loss of MHC I expression, and expression of immune inhibitors such as PD-L1 [141,142,143] or suppressive mediators such as IL-10 [144], TGF-β [145], and TRAIL decoy receptors [146,147]. Recruitment and activation of immune-suppressing cells such as Tregs also contribute to escape [148].
4.1. Effect of TEVs on Dendritic Cells
Maturation of DCs requires inflammation-related stimuli which stimulate the expression of co-stimulatory molecules such as CD86, CD80, and CD40. TEVs can modify or block the differentiation of immature myeloid cells (IMC) to DC or divert the DCs maturation from IMC to MDSC or M2 macrophage by interacting with bone marrow IMC and inducing the production of IL-6, and decreasing expression of CD83 and CD86, as reported for breast cancer, murine mammary adenocarcinoma, and melanoma [149,150]. TEVs also can disrupt DC maturation and T-cell immune response with HLA-G-associated mechanisms in renal cancer [133] (Table 1). Some vesicular proteins such as MALAT1 directly interact with DCs and induce DC autophagy, which decreases DC-mediated T-cell activation [151]. Furthermore, TEV-treated DCs were ineffective at inducing CD4+ T-cell proliferation and activation but promoted differentiation into Treg [152]. TEVs fatty acids can create immunologically dysfunctional DCs by increasing intracellular lipid content by activating the peroxisome proliferator-activated receptor (PPAR) resulting in extra fatty acid oxidation (FAO) which shifts the DCs’ metabolism toward oxidative phosphorylation of mitochondria and the disruption of the function of DCs [153,154,155]. It was reported that human prostate cancer-derived extracellular vesicles purified from cultured cells contained PGE2 and triggered the expression of CD73 and CD39 on DCs in vitro, resulting in the generation of adenosine from ATP and inhibition of TNF-α and IL-12-production which reduced T-cell activation [156].
Table 1. Effect of the tumor-derived extracellular vesicles on Dendritic cells.
| Cancer Type. | Cellular Source | Vesicular Cargo | The Main Result | Refs. |
|---|---|---|---|---|
| Prostate cancer | DU145 | PGE2 | Triggered the expression of CD73 and then CD39 on DCs, resulting in inhibition of TNFα- and IL-12-production via an ATP-dependent manner | [156] |
| NSCLC | Blood samples from NSCLC patients | Galectin-9 and Tim-3 | Interacted with TIM-3 on DCs | [157] |
| Renal cancer | CD105+ CSCs CD105− TCs |
HLA-G | Disrupted maturation of DCs and T-cell immune responses | [133] |
| Glioblastoma | CSF samples from glioma patients GL261 U87MG U118 MG |
Galectin-9 | Inhibited antigen recognition, processing, and presentation by interacting with TIM-3 on DCs | [142] |
| Ascites of glioma patients | PD-L1 | Impaired DCs maturation via formation of immunosuppressive monocytes | [77] | |
| Blood samples from glioma patients GSC20 GSC267 GSC17 MEC-1 |
Vesicular cargo | Skewed monocytes toward an immune suppressive phenotype and induced programmed PD-L1 expression on monocytes through STAT3 phosphorylation and TLR7-dependent manner | [33,158] | |
| Melanoma | SKMEL28 A375 C32TG |
S100, A8/A9 | Inhibited DCs maturation and reduced expression of CD83, CD86, Th1 polarizing chemokines (such as Flt3L, IL-15), and migration chemokines (MIP-1α and MIP-1β) | [150] |
| lymphatic fluid sample of melanoma patients ATCC |
S100A9 | Inhibited DCs maturation and prepared metastatic niche in lymph nodes | [159] | |
| B16-F0 | TGF-β1 | Increased mRNA levels of IL-4 and TGF-β1 which inhibited DCs’ maturation | [160] | |
| Blood samples from melanoma patients B16-F0 |
HSP72 and HSP105 | Induced secretion of IL-6 from DCs via TLR4- and TLR2-dependent manner activating STAT3-dependent MMP 9 activity | [161] | |
| lymphocytic leukemia | Blood samples from CLL patients | S100A8/A9 | CD83, CD86, IL-12, and IL-15 expressions were all downregulated via activating the NFκB pathway | [162,163] |
| lung carcinoma | LLC | PD-L1 | Myeloid precursor cells were unable to differentiate into CD11c+ DCs in the presence of vesicular PD-L1 and resulted in DCs death | [152] |
| LLC A549 |
MALAT1 | Inhibited DC function and T-cell proliferation and increased DC autophagy via AKT/mTOR Pathway | [151] | |
| Breast cancer | MDA-MB-231 TS/A |
Vesicular cargo | Inhibited the development of myeloid precursor cells into DCs by increasing IL-6 production and reducing CD83 and CD86 expression | [149] |
| 4T1 | PD-L1 | Myeloid precursor cells were unable to differentiate into CD11c+ DCs in the presence of vesicular PD-L1 and resulted in DC death | [152] | |
| Blood samples from melanoma patients 4T1 |
HSP72 and HSP105 | Promoted DCs to IL-6 secretion in a TLR2- and TLR4-dependent manner which activated STAT3-dependent MMP 9 activity | [161] |
HSP72 and HSP105 on the membrane of TEVs interact with TLR2 and TLR4 on DCs which induced IL-6 secretion by DCs that increased STAT3-dependent MMP-9 transcription activity in cancer cells resulting in tumor invasion [161]. Galectin-9 on glioblastoma-derived EVs binds to the TIM3 DCs receptor and inhibits antigen presentation by DCs, leading to disrupted antitumor immune responses of cytotoxic T cells [142]. Important DC receptors such as Tim-3 and galectin-9 [157] and SIRPα as the ligand for CD47 were up-regulated on the tumor cells’ membranes and derived TEV [143,164]. TLR4 on the DCs decreased after treatment with pancreatic cancer-derived vesicular miR-203 resulting in reduced expression of cytokines such as TNF-α and IL-12, subsequently reducing DC maturation and Th1 differentiation [125]. Besides the vesicular proteins, vesicular miRs also affect DC’s function. For example, miR-212-3p transferred to DCs by pancreatic cancer-derived extracellular vesicles suppressed regulatory factor X-associated protein (RFXAP), decreased MHC II expression, and reduced antigen presentation by DCs [165]. Table 1 summarizes reports of TEV impacts on DC.
4.2. Effect of TEVs on T Cells
TEVs have a broad array of mechanisms by which they impact T cells. TEVs modify antitumor response by reducing T-cell viability, proliferation, and effector activities [166,167,168]. TEVs can disrupt T-cell effector function indirectly by blocking APC maturation [142,151,152] or directly by inhibiting activated CD8+ T-cell function, inducing CD8+ T-cell death through pro-apoptotic molecules (galectin-group proteins and FasL), promoting Treg expansion, and inducing T-cell exhaustion [169,170]. PD-L1 enriched glioblastoma-derived EVs perhaps surprisingly suppress monocytes rather than T-cells [77]. Nasopharyngeal carcinoma-derived vesicular galectin-9 induced apoptosis in CD4+ T cells via interaction with Tim-3 [171], as well as impairing T-cell function by interaction with TIM3 receptor on DCs in glioblastoma [142]. TEVs can carry pro-apoptotic Bax that induces apoptosis in CD8+T cells [172] and downregulates JAK3 expression which blocks CD8+ T-cell activation [167,173]. In Treg cell activation, both CD45 negative and positive EVs derived from plasma in head and neck cancer induced Treg differentiation of CD4 cells, but CD45(-) EVs also reduced CD8+ T-cell activation due to their higher adenosine concentrations [174]. EVs generated from multiple myeloma reduced the viability of CD4+ T cells and boosted the proliferation of Treg cells [175].
Vesicular PD-L1 promotes CD8+ T-cell apoptosis via PD-1/PD-L1 and PD-L1/CD80 signaling pathways [176], blocks T-cell activation in the draining lymph node in TRAMP- C2 prostate cancer mouse model [177,178], and reduces the proliferation of CD8+ T cells by decreasing IL- 2 and IFN-γ in the TME [136]. FasL on the TEVs decreased T-cell receptor (TCR) and CD3ζ expression in T cells leading to T-cell apoptosis [179], and melanoma-derived vesicular TNF downregulates TCR via redox signaling in T cells [180].
Pancreatic cancer cell EVs can stimulate p38 MAP kinase signaling in T lymphocytes that causes ER stress, which triggers the PERK–eIF2–ATF4–CHOP signaling cascade resulting in T-cell death [181]. Vesicular microRNAs in the serum of patients with nasopharyngeal carcinoma influenced T-cell differentiation and activation through suppression of the MAPK1 signaling pathway [182], while EVs with a high amount of miR-24–3 reduced CD4+ and CD8+ T-cell proliferation by targeting FGF11 [183]. In addition, mesothelioma cells’ EVs carrying TGF-β decreased proliferative response to IL-2 in T effector cells, but not in T-reg cells [184].
Vesicular galectin-1 plays a role in the induction of T-cell suppression [185]. TEVs also can induce T-cell exhaustion, by carrying inhibitory molecules, including PD-L1, CTLA- 4, TIM3, LAG3, and TIGIT [186,187]. miR-146a-5p and 14-3-3ζ in HCC-derived EVs induced T-cell exhaustion via activating M2-macrophages by inhibiting transcription factor SALL4 [30,188]. EVs carrying circRNA-002178 from patients’ serum with lung adenocarcinoma could boost PD-L1 production by sponging miR-34 in cancer cells, leading to CD8+T-cell exhaustion in vitro [132].
In addition, cancer patients’ plasma TEVs can prevent the activation of Th1 and Th17 lymphocytes and change them to immunosuppressive Treg phenotype cells [167,182]. The mutant KRAS gene is involved in the NSCLC-generated EVs-mediated transition of naive CD4+ T cells towards a FoxP3+ T-reg phenotype in a cytokine-independent manner in an NSCLC xenograft mouse model [189]. Table 2 summarizes reports on TEV suppressive effects on T cells.
Table 2. Effect of the tumor-derived extracellular vesicles on T cells.
| Cancer Type | Cellular Source | Vesicular Cargo | Mechanism of Action | Refs. |
|---|---|---|---|---|
| Ovarian cancer | Ascites of ovarian patients OVCAR3 SKOV3 AD10 |
TGF-β1, IL-10 |
Increased IL-10, FasL, TGF-β1, CTLA-4, which promoted Treg proliferation, suppressor activity, and Treg cell survival. | [190] |
| Blood samples from ovarian patients Ascites of ovarian patients OVCAR-3 AD10 A2780 Skov3 CaOv-3 MDAH2774 OvCa-14 OVP-10 |
Arginase-1 | Inhibited antigen-specific T-cell proliferation | [191] | |
| Prostate cancer | Pleural fluid samples of malignant pleural mesothelioma patients DU145 PC3 |
PGE2 | T-cell inhibition was mediated through the adenosine A2A receptor | [192] |
| DU145 PC3 |
TGF-β1 | Skewed IL-2 responses in T cells and suppressed cytotoxicity | [184] | |
| Melanoma | Blood samples from melanoma patients Blood samples from melanoma tumor-bearing mice WM1552C WM35 WM793 WM902B UACC-903 1205Lu WM9 WM164 |
PD-L1 | Suppressed the function of CD8 T cells | [136] |
| Colorectal cancer | Blood samples from colorectal patients SW403 CRC28462 1869col |
FasL, TRAIL | Induced T-cell apoptosis | [168] |
| DLD-1 WiDr |
TGF-β1 | Induced differentiation of T cells to Treg-like cells via the TGF-β pathway while inactivating the SAPK signaling pathway | [193] | |
| Caco-2 | Galectin- 1 | Induced suppressor phenotype in human CD8+ T cells | [185] | |
| Head and neck cancer |
Tu167 SCC0209 HN60 |
Galectin- 1 | Induced suppressor phenotype in human CD8+ T cells | [185] |
| Blood samples from HNSCC patients | Vesicular cargo | Induced apoptosis in CD8+ T cells by converting CD4+ T cells to Treg | [174] | |
| Glioblastoma | Blood samples from glioma patients UPN933 E3-2 E6-5 |
Vesicular cargo | Deactivated T cells by FasL-dependent mechanisms and inhibit secretion of IL-2 | [194] |
| Nasopharyngeal cancer (NPC) | Blood samples from NPC patients Blood samples from NPC tumor-bearing mice C15 C17 |
Galectin- 9 | Induced huge apoptosis in T cells via membrane receptor Tim-3 | [171] |
| Blood samples from NPC patients C15 C17 |
CCL20 | Facilitated Treg recruitment and expansion that increased secretion of immunosuppressive cytokines (IL10, TGFB1) | [195] | |
| Blood samples from NPC patients TW03 C666 CNE2 |
miR- 24–3p | Blocked T-cell proliferation and Th1 and Th17 differentiation and promoted Treg induction via dephosphorylating ERK, STAT1, and STAT3 by reducing IL-2, IFNγ, and IL-17 secretion and phosphorylating STAT5 with increasing IL-6, IL-1β, and IL-10 secretion | [182,183] | |
| Oral squamous cell carcinoma(OSCC) | SCC-9 SCC-4 CAL-27 |
HSP70 | Altered development and cytotoxicity of T cells in an HSP70-dependent way via miR-21/PTEN/PD-L1 regulatory pathway | [170] |
| Blood samples from OSCC patients PCI-13 |
FasL | Induced apoptotic pathways in T cells through triggering caspase-3 cleavage, the release of cytochrome c that led to disrupting mitochondrial membrane, and decreased TCR-ζ chain production | [172] | |
| Breast cancer | MCF7 | CD73, CD39 | Inhibited T cells via the adenosine A2A receptor | [192] |
| BT-474 MDA-MB-231 |
TGF-β1 | Suppressed T-cell proliferation | [196] | |
| Lung cancer | Blood samples from lung cancer patients A549 PC9 95D |
circRNA- 002178 |
Enhanced PDL1 expression led to induced T-cell exhaustion | [132] |
| Hepatocellular Carcinoma (HCC) |
Blood samples from HCC patients MHCC97H |
14- 3- 3ζ | Inhibited the functions of T cells against cancer in the HCC microenvironment | [188] |
| Hepa1-6 H22 |
SALL4/miR-146a- 5p | T cells were exhausted by reducing IFN-γ and TNF-α expression while increasing the expression of inhibitory receptors such as PD-1 and CTLA-4 | [34] | |
| Pancreatic cancer | BxPC-3 tdTomato-BxPC-3 |
Vesicular cargo | Induced ER stress-mediated apoptosis via activating the p38 MAP kinase signaling | [181] |
4.3. Effect of TEVs on NK Cells
NK cells play an important role in cancer immunosurveillance by expressing death-inducing ligands such as FasL, TRAIL and JAK/STAT pathway [197,198]. However, like most immune cells, the activation of NK cells is controlled by a complex balance of activating and inhibiting signals. Tumor cells trigger several activating receptors, such as NKG2D, natural cytotoxicity receptors (NCRs), and DNAX accessory molecule-1 (DNAM-1/CD226) [199]. Vesicular NKG2D, TGF-β, and MICA*008 suppress or downregulate the expression of NKG2D in both NK and CD8+ T cells resulting in decreasing cytotoxicity of these cells by reducing the expression of cytotoxic molecules [200,201,202,203,204,205,206].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15010082
This entry is offline, you can click here to edit this entry!