1. Hyperglycemia and Diabetic Nephropathy
It is known that hyperglycemia induces the increase of the osmotic pressure of biological fluids due to a decrease in the glucose absorption rate of cells [
14]. As a result, diuresis develops, which means loss of water and salts from kidneys, water exhaustion, and deficits of ions. On the other hand, the intensity of non-enzymatic glycosylation of proteins and lipids increases in these conditions [
15].
One of the most typical diabetic complications is diabetic nephropathy (DN), which usually decreases the life quality, brings invalidation, and is the largest single cause of end-stage renal disease [
16,
17].
DN is characterized by the development of sclerosis of the renal glomeruli, leading to impaired renal function, primarily the filtration function of the kidneys, and the development of chronic renal failure [
18].
Following the modern classification [
19] DN has three stages of development: microalbuminuria (MAU); proteinuria (PU); and chronic kidney disease (CKD).
MAU is recognized as an early predictor of nephropathy [
20]. In this stage, the albumin excretion is 30 and 300 mg/day; in patients with T2DM, high blood pressure can be observed, and it is important to point out that it will be possible to recover kidney functions if treatment starts on time [
21].
PU is known as a visible or real stage of DN. In this stage, albumin in urea is above 300 mg/day, kidney glomerular filtration rate (GFR) decreases, and stable high blood pressure is observed. Treatment is based on angiotensin change enzyme (ACE) inhibition or angiotensin receptor blocking [
22].
CKD is a terminal stage of DN. This stage is typically characterized by a high level of blood creatinine and urea; GFR is less than 60 mL/min, and hypertension progresses. When GFR becomes less than 15 mL/min, kidney therapy is necessary (hemodialysis, dialysis, and even kidney transplantation) [
17,
23].
An understanding of the mechanisms of DN is a necessity and there are a lot of studies on this path [
24,
25]. A large amount of literature data are available regarding the polyethiological nature of DN. The most attention is given to the genetic, metabolic, and vascular factors [
26,
27].
There is an accepted fact that in the basis of pathological mechanisms of DN development, three factors are underlying: hyperglycemia and dyslipidemia; intraglomerular hypertension; and arterial hypertonia, where oxidative stress plays a crucial role [
27].
Hyperglycemia is an initial metabolic “button” of the DN. On the one hand, the high-level glucose leads to the non-enzymatic glycosylation of glomerular proteins and lipids and damages glomerular vesicles. On the other hand, glucose increases vascular permeability due to its direct toxic effect on kidney tissue [
27]. As a result, the tone of the adductor blood vessel decreases, and it expands, while the efferent vessel saves its tension due to the effect of angiotensin II [
28]. These changes bring renal hypertension and, as a sequel, glomerulosclerosis develops. Arterial hypertonia, which is typical of T2DM, contributes to the progression of glomerulosclerosis as well [
2].
2. T2DM and Inflammation
Despite diabetes not being thought to be an immune disease, there are many studies pointing to the role of inflammation in T2DM [
29]. In this context, it is important to note the influence of TNFα, IL1, and IL6 in the pathophysiological mechanisms of IR [
30]. Animal experiments have shown increased levels of TNFα in adipose tissue in obesity; its neutralization restored glucose uptake by peripheral tissues [
31]. These results were also confirmed for humans, where the TNFα level correlates with insulin resistance (IR) and decreases with weight loss [
32]. Finally, a mechanism has been demonstrated whereby inflammatory cytokines can interrupt signaling from the insulin receptor internally [
33]. All these data formed the basis for the creation of the concept of the relationship between inflammation and IR.
T2DM is an obesity-related metabolic syndrome with the sustained activation of the NLRP3 inflammasome, which is a critical component of the innate immune system mediating caspase-1 activation and the secretion of proinflammatory cytokines IL-1β/IL-18. The activation of the NLRP3 inflammasome is linked with inflammatory disorders such are Alzheimer’s disease, diabetes, and atherosclerosis. The NLRP3 inflammasome is activated by different factors and diverse molecular and cellular events such as ionic flux, production of ROS, mitochondrial dysfunction, and lysosomal damage [
30,
34]. It is important to point out that T2DM is accompanied by oxidative stress-induced ROS production, which could result in NF-κB activation and the transcription of NLRP3 and, as a result, the activation of pro-IL-1β and pro-IL-18 [
34,
35] (
Figure 1).
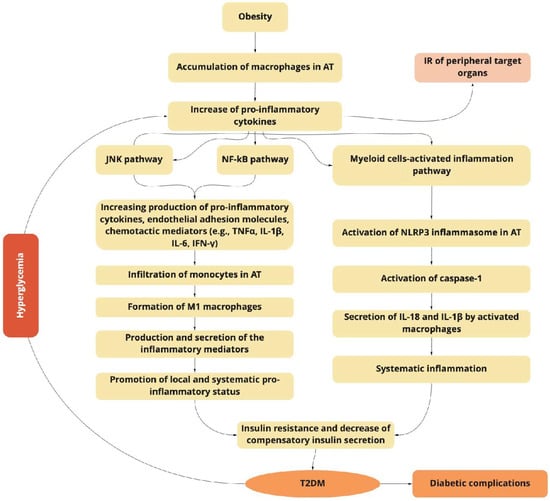
Figure 1. The obesity-related inflammation as a predictor of type 2 diabetes mellitus (T2DM) induction and progression.
The obesity-related accumulation of macrophages in adipose tissue (AT) activates the JNK and NF-κB signaling pathways, increasing the production of pro-inflammatory cytokines, endothelial adhesion molecules, and chemotactic mediators, which contribute to the infiltration of monocytes, and the formation of pro-inflammatory M1 macrophages. Macrophage-secreted diverse inflammatory mediators promote the local and systemic pro-inflammatory state and induce insulin resistance of targeted tissues. The NF-κB inflammatory pathway results in the increased expression of several NF-κB target genes (e.g., IL-6, TNFα, IFN-γ, and IL-1β), exacerbating the IR progression. Myeloid cells activate the inflammasome pathway connected with the macrophages and other innate immune cells. The last ones initiate inflammatory responses by detecting pathogen- or danger-associated molecular patterns by pattern-recognition receptors such as NLRP3, which plays a key role in the obesity-specific chronic inflammation and progression of IR. The activation of caspase-1 mediates the secretion of IL-1β and IL-18 by macrophages. The increasing production of IL-1β in pancreatic islets and insulin-sensitive tissues is associated with T2DM. IL-18 enhances the maturation of T- and NK-cells, and increases the production of diverse pro-inflammatory cytokines, exacerbating obesity-induced systemic inflammation.
Activation of the NLRP3 inflammasome in adipose-tissue-infiltrating macrophages brings metabolic inflammation, which in its turn aggravates the inflammation in insulin-sensitive tissues [
36]. During β-cell failure, activation of NLRP3 could be released via alternative mechanisms. Initially, due to hyperglycemia, the β-cell-derived mitochondrial ROS is produced, which leads to the dissociation of thioredoxin-interacting protein from thioredoxin and then the activation of NLRP3 [
37]. Furthermore, continuous hyperinsulinemia brings the accumulation of a large amount of IAPP around the islet cells, which, in its turn, specifically, activates the NLRP3 inflammasome [
38]. Animal studies using mice with beta-cell-specific overexpression of IAPP revealed a strong induction of IL-1β in pancreatic macrophages [
39].
3. Inflammation and DN
A wide range of changes including hemodynamic and metabolic disorders, the upregulation of the renin-angiotensin system (RAS), oxidative stress, and fibrosis are the main characteristics of DN [
40]. All these alterations together bring the increase of systemic and intraglomerular pressure, and the development of diverse symptoms associated with the development of kidney failure, such as glomerular hypertrophy, and decreasing glomerular filtration [
23]. Recent advances indicate that kidney complications in DM are not only a result of alterations in hemodynamic and metabolic factors, but a complex and multifactorial process (
Figure 1 and
Figure 2) [
26].
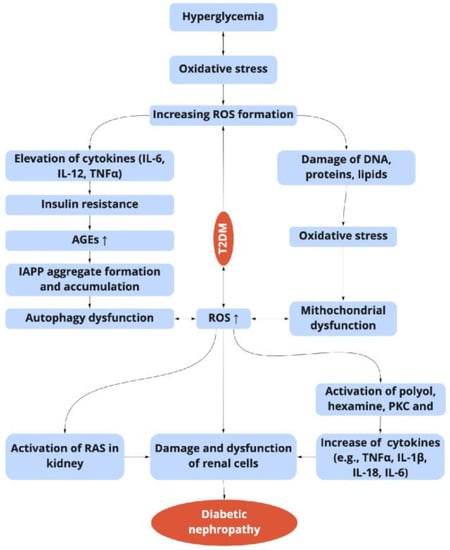
Figure 2. Relationship between the hyperglycemia-induced oxidative stress, inflammation, and diabetic nephropathy in T2DM. ROS: reactive oxygen species; TNF-α: tumor necrosis factor-α; NF-κB: nuclear transcription factor-κB; AGEs: advanced glycation end products; PKC: protein kinase; IAPP: islet amyloid polypeptide; RAS: renin-angiotensin system.
Following the last studies, inflammation plays a key role in the pathophysiological mechanisms of DN [
41]. In this context, it is necessary to discuss the “weight” of pro-inflammatory pathways and molecules in the progress of renal damage during the development of the disease. A large spectrum of pro-inflammatory molecules and pathways participate in the pathophysiological processes of diabetic nephropathy, including pro-inflammatory cytokines, chemokines, their receptors, adhesion molecules, and transcription factors [
42]. The increase of pro-inflammation cytokines in the blood of patients has been noted, and a direct correlation has been found between DN progression, albuminuria, and the increase in the concentration of the pointed cytokines [
43,
44].
Numerous inflammatory parameters foretold the initiation and progression of DN [
45]. Inflammatory cytokines play a binary role. They regulate the immune response and play key roles as basic promoting elements of injury. The elevated concentrations of these elements in T2DM patients trigger microvascular complications, particularly the development of nephropathy [
46]. Both the circulating pro-inflammatory cytokines and those synthesized and secreted by inflammatory cells in the kidney tissue are increased in patients with DN, and, simultaneously, are in direct correlation with urinary albumin excretion (UAE) levels and clinical markers of glomerular and tubulointerstitial damage [
47].
The pathophysiological changes observed at various levels of DN contribute to the development and progression of kidney damage [
48].
Inflammatory mechanisms are crucial in the DN pathophysiology and explain how metabolic and hemodynamic disorders in DM patients translate to structural and functional kidney injuries.
Thus, IL-18 is a proinflammatory cytokine synthesized by renal tubular cells and by infiltrated monocytes, macrophages, and T-cells as well [
49]. These cytokine levels are elevated in the serum and urine of patients with DN. Significant and direct correlations are observed between IL-18 and UAE levels, and consequently, the evolution of albuminuria [
50]. The level of IL-18 could be used as an early marker of renal dysfunction in T2DM patients. Additionally, the levels of IL18 in serum correlate with the level of β2-microglobulin in urine, which is a marker of tubular dysfunction [
50]. It is important to note that IL18 modulates the synthesis of IL-1, TNF-α, and interferon γ (IFN-γ), which in its turn activates chemokine receptors in mesangial cells. Moreover, IL8 enhances the expression of intercellular adhesion molecule 1 (ICAM-1) [
50] and facilities endothelial cell apoptosis [
37]. In addition to infiltrating cells, kidney tubular cells of patients with DN express raised levels of IL-18 as well, which is connected with the activation of the mitogen-activated protein kinase (MAPK) pathways by transforming growth factor (TGF) β [
51].
Serum and urinary levels of TNF-α elevate in patients with DN in parallel with the progression of renal injuries, which may point to a relationship with the development and progression of renal deficiency [
52]. It was shown that the increasing level of TNF-α has a cytotoxic effect on the renal tissue, while the inhibition of TNF-α improves markers of glomerular and tubulointerstitial injuries in DN patients [
53]. Some investigations are stating that the increasing levels of TNF-α in kidney glomeruli and tubules are directly and independently associated with the UAE [
45,
54].
In the pathogenesis of DN, IL-1 is also involved [
55]. This cytokine triggers the synthesis of prostaglandin E and the release of phospholipase A2. The latter play a critical role in the progression of intraglomerular hemodynamic abnormalities. Due to the influence of IL-1, the permeability of vascular endothelial cells increases as well [
50].
In animal DN models, the activation of IL-1 expression is revealed in many types of renal cells [
56,
57]. The relationship between IL1 activity and the expression of intercellular adhesion molecule 1 (ICAM-1), as well as the vascular cell adhesion molecule-1 (VCAM-1), and the endothelial-leukocyte adhesion molecule-1 (ELAM-1), have been shown [
58]. Rat kidney mesangial cells produce prostaglandin E2 after incubation with recombinant IL-1 in response to angiotensin II, which might result in the appearance of abnormalities in intraglomerular hemodynamics [
50]. IL-1 is associated with the secretion of hyaluronan in the proximal tubule, which is related to the progression of hypercellularity [
59].
IL-6 is another cytokine that is involved in DN progression due to its pleiotropic effects [
60]. The increase of IL-6 enhances proliferation of the extracellular matrix and influences vascular permeability which in its turn brings to DN. IL-6 participates in facilitating the neutrophil infiltration of the tubule-interstitium, acts on extracellular matrix dynamics, and contributes to overall kidney hypertrophy, thickening the renal glomeruli and podocyte hypertrophy, correlating with albuminuria [
61].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27249035