Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Hepatocellular carcinoma (HCC) is a global healthcare challenge, which affects more than 815,000 new cases every year. Activated hepatic stellate cells (aHSCs) remain the principal cells that drive HCC onset and growth. aHSCs suppress the anti-tumor immune response through interaction with different immune cells. They also increase the deposition of the extracellular matrix proteins, challenging the reversion of fibrosis and increasing HCC growth and metastasis.
- hepatocellular carcinoma
- hepatic stellate cells
- fibrosis regression
1. Introduction
Liver cancer is a global health problem, with an estimated increase of 32% by 2040 [1]. Representing 90% of liver cancers, hepatocellular carcinoma (HCC) causes 700,000 deaths annually.
Although HCC pathogenesis is complex and varies depending on underlying etiology, the usual background setting for HCC is liver injury, chronic inflammation, irreversible fibrosis, and cirrhosis [2]. In fact, 80–90% of HCC develops in the fibrotic or cirrhotic liver [3]. Hepatic stellate cells (HSCs) play a key role in this sequence of events, contributing mainly to liver fibrosis and cirrhosis. They are liver-specific mesenchymal cells, which are located in the perisinusoidal space in contact with different cell types [4]. In a healthy liver, HSCs exist in a quiescent non-proliferative state as an important source of paracrine, autocrine, and chemoattractant factors to maintain hepatic homeostasis [5]. Quiescent HSCs are very sensitive to extracellular pro-fibrotic signals [6] and contain numerous vitamin A lipid droplets, which are essential for the proper function of the immune system [7]. When toxins or viruses injure the liver, damaged hepatocytes and immune cells secrete signals, which could activate HSCs into myofibroblast-like cells [8]. Activated HSCs produce an extracellular matrix (ECM) at the site of injury as a temporary protective scar to prevent further damage, initiating the first steps of fibrosis [8][9]. Long-acting agents maintain the activation of HSCs, increasing their capabilities for proliferation and migration [10]. Activated HSCs produce more ECM, leading to chronic fibrosis and cirrhosis and eventually to HCC (Figure 1) [6]. Despite significant advances in the treatment of HCC, drug-resistance is a critical obstacle [11], and the 5-year survival rate is low (5–14%) [12], but survival rates of greater than 20% have been reached in some regions [13]. Therefore, there is a potential to increase survival rates. Given the fact that available therapies can activate HSCs, synchronous targeting activated hepatic stellate cells (aHSCs) may be beneficial for patients [14].
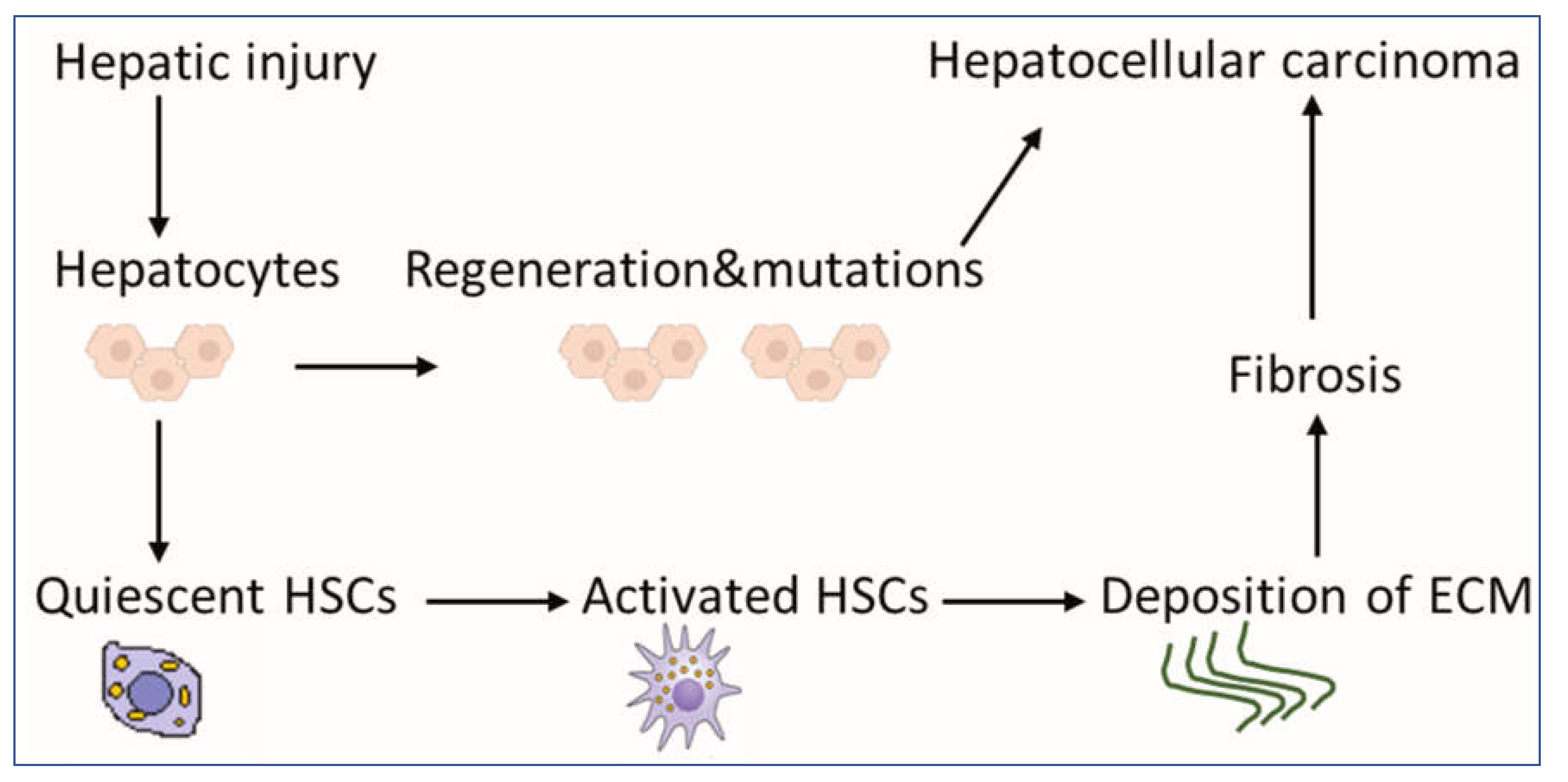
Figure 1. Scheme for the contribution of HSCs to liver pathology. HSCs exist in a quiescent state, containing numerous vitamin A lipid droplets. When the liver is injured, damaged hepatocytes mediate HSC activation, which could produce a large amount of ECM, leading to fibrosis as an indirect mechanism of HCC. Mutations during the regeneration of hepatocytes may lead directly to the development of HCC. Abbreviations: HSCs: hepatic stellate cells, HCC: hepatocellular carcinoma and ECM: extracellular matrix.
2. The Role of aHSCs in HCC
2.1. The Suppression of the Antitumor Immune Response by aHSCs
In HCC, aHSCs receive signals from individual immune cells, and, in turn, they produce soluble mediators, acting on surrounding immune cells [15]. HSC mediators could orchestrate both innate and adaptive immunity, resulting in an immunosuppressive tumor microenvironment [7]. Reduction of antitumor responses was shown in immunocompetent mice after co-transplantation of HSCs and HCC cells [16]. Such cotransplantation of HSCs inhibited systemically lymphocyte infiltration, which promoted tumor cell proliferation and, therefore, HCC growth; the size of the tumors was HSC dose-dependent [16]. Previous experiments addressing HCC–HSC interactions were performed on immunodeficient mice, and, therefore, the effect of HSC on the immune system was not investigated. Using immunocompetent mice, the authors were able to define HSC-immune interactions in HCC [16].
The antigen-directed cytotoxicity of T lymphocytes (TLs) boosts the immune response against cancer [17]. Activation and proliferation of TL in tumor tissue, predominantly CD8+ and CD4+ T lymphocytes, can control HCC progression [18]. HSCs can exert their immunomodulatory activities by downregulating the number and function of CD4+ and CD8+ TLs [19]. Contrary to quiescent HSCs, aHSCs in mice and humans expressed programmed death-ligand 1 (PD-L1) to inhibit TL responses [7]. PD-L1 expressed by HSCs can induce TL apoptosis, attenuate TL infiltration, and suppress TL-mediated cytotoxicity, therefore inhibiting TL responses and enabling tumor cells to escape the host immune response [20]. In addition, HSCs may prevent the local stimulation of naive TLs [21]. In Hepa1–6 cells, activated HSCs induced the death of activated TLs and reduced the cytotoxicity of cancer-specific TLs, which resulted in the increased proliferation and migration of cancer cells [22]. More investigation of the role of aHSCs in the apoptosis of TLs in HCC patients is needed.
aHSCs also induce expansion of two suppressive immune cell populations; myeloid-derived suppressor cells (MDSCs) [14] and T helper 17 (Th17) cells, a subset of CD4+ effector T cells [23]. MDSCs play a pivotal negative role in the immune response through the inhibition of cytotoxic T cells and recruitment of regulatory T cells, which results in tumor progression [24]. HSCs induce MDSC accumulation in the tumor tissue by the stimulation of the COX2–PGE2–EP4 pathway [25]. Inhibition of this pathway in murine orthotopic HCC models downregulated MDSCs and HCC growth [25]. Immunosuppressive functions of Th17 cells may contribute to HCC progression [26]. IL-17A produced by Th17 could increase cancer cell motility via the activation of the nuclear factor-kB (NF-kB) transcript factor, increasing HCC metastasis [27]. Culturing CD4+ cells with HSCs (extracted from hepatitis B virus-related fibrotic liver tissue) increased the percentages of Th17 cells [23]. HSCs may secrete high levels of interleukin-6 as a critical initiator of Th17 expansion and tumor necrosis factor-α as a key regulator of Th17 differentiation [28]. Interestingly, previous data indicated suppression of Th17 differentiation by mouse HSCs [29]. Critical evaluation of Th17-HSCs interactions could be addressed in appropriate mouse models of HCC.
Macrophages polarize in the liver with strong plasticity into pro-inflammatory M1 or anti-inflammatory M2 in response to local signals from the tumor microenvironment [30][31]. M1 macrophages are thought to be tumoricidal, while M2 macrophages are usually believed to promote tumorigenesis and tumor progression [32]. M2 macrophages in HCC promote the invasion and migration of tumor cells [33]. M2 macrophage-derived CCL22 was proven to enhance tumor migration through the activation of epithelial–mesenchymal transition [34]. aHSCs recruited CCL2/CCR2 pathway in HCC cell lines to stimulate M2 phenotypic transformation [35]. M2 macrophage polarization could lead to the progression of HCC [36].
Natural killer (NK) cells defend the body against tumors by engaging death-inducing receptors to stimulate cancer cell apoptosis. HCC patients with low intratumoral NK cells infiltration have shorter disease-free survival [37]. In animal models of fibrosis, transforming growth factor-β secreted by HSC could inhibit NK cell function [38]. On the other hand, NK cells could induce apoptosis of aHSCs in hepatitis C virus-infected patients [39] and mouse models of fibrosis [40]. Studies on the interaction between aHSCs and NK cells in HCC models should be investigated. Dendritic cells (DCs) can activate antitumor immunity by priming TL against cancer-progression-associated antigens. HSCs induce the expression of dendritic-cell-derived immunoglobulin receptor 2 (DIgR2), which inhibits DC-induced antigen-specific TL responses [41]. DIgR2 was shown to bind to the receptor in TLs, suppressing TL proliferation, cytokine production, and cytotoxic TL activity [41]. Co-culturing of tumor-HSCs (isolated from the tumor) to DCs induced the expression of DIgR2, in contrast to quiescent HSCs, which had no significant effect on DIgR2 expression. Considering quiescent HSCs in such studies boosts the role of activated HSCs in HCC [41].
Although the role of immune system in liver cancer is complex [42], the overall role of aHSCs in immune regulation is pro-oncogenic [16]. Exploring the interaction between different immune cells and HSCs in established HCC models could highlight the main targets to improve immune surveillance against HCC.
2.2. HSCs Upregulate the Deposition of ECM for the Development of Fibrosis and HCC
Under normal conditions, the rate of ECM production in the liver equals that of its degradation, resulting in no net accumulation of the matrix. Fibrogenesis occurs when there is an imbalance between ECM production and degradation [43], resulting in the impairment of liver functions, which may eventually lead to cirrhosis and HCC [44]. Fibrosis and cirrhosis are reported clinically as reversible processes [45]. Reversibility of liver fibrosis depends mainly on the degradation of ECM [46]. aHSCs are able to increase matrix protein synthesis, which might lead to the irreversibility of fibrosis and favor progress and metastasis of HCC (Figure 2).
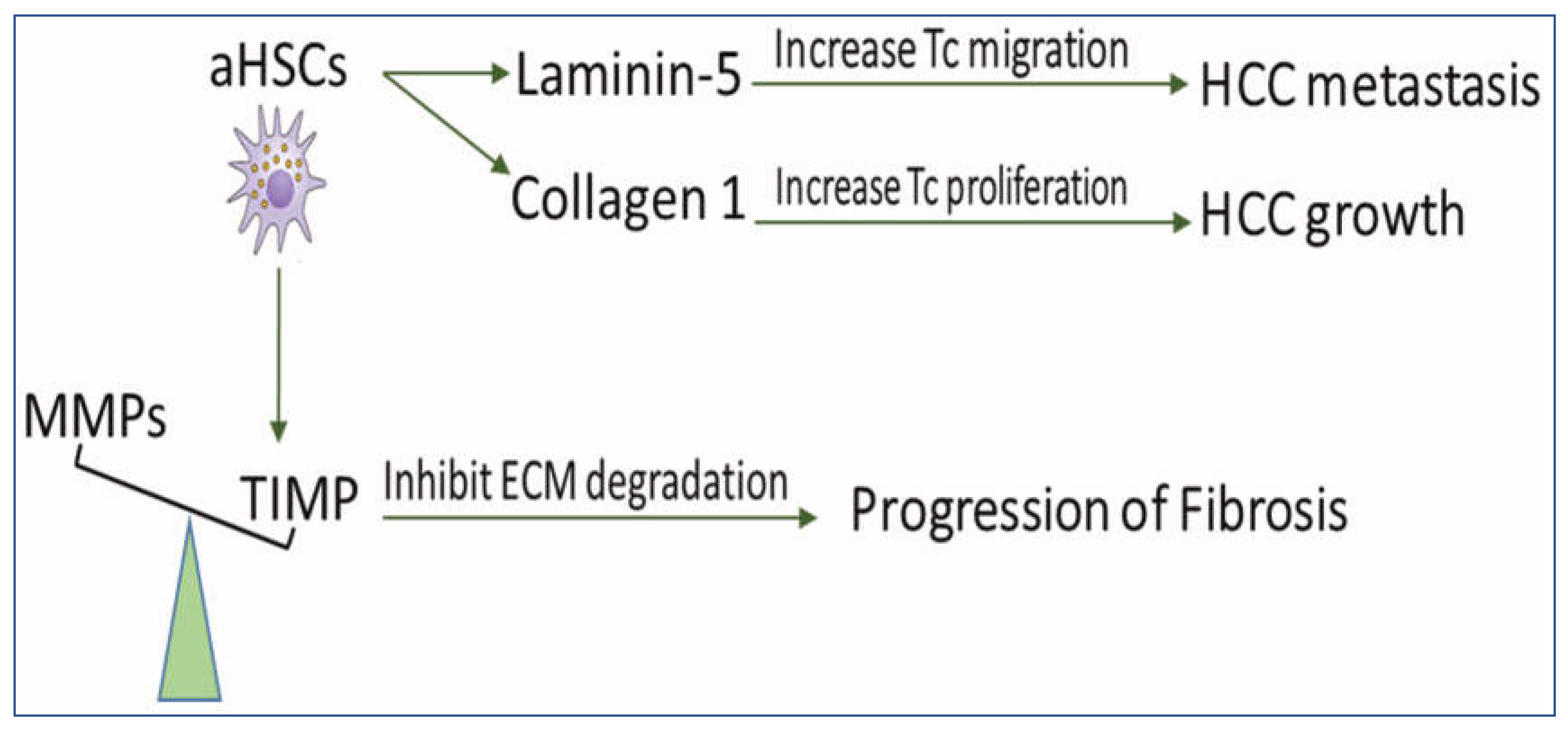
Figure 2. The scheme demonstrates the role of HSCs in disturbing ECM balance for the development of fibrosis and HCC. aHSCs express TIMP glycoproteins which inhibit MMP-mediated degradation of ECM and, therefore, prevent fibrosis regression. They also secrete collagen I, which increases tumor cell proliferation and produces Ln-5, which promotes tumor cell migration. MMPs: matrix metalloproteinases, TIMPs: tissue inhibitors of MMP, aHSCs: activated hepatic stellate cells, HCC: hepatocellular carcinoma, ECM: extracellular matrix, and TC: tumor cell.
The reversibility of fibrosis and cirrhosis is dependent on the activity of matrix metalloproteinases (MMPs) [47]. MMPs are a group of enzymes involved in the degradation of ECM-proteins, which are blocked by tissue inhibitors of MMP (TIMPs). It has been reported that prolonged expression of TIMPs, even after withdrawal of fibrogenic factors, slows the regression of liver fibrosis [47]. In a rat model of regressed liver fibrosis, the reversibility of fibrosis was increased in parallel with a marked decrease in TIMP expression [48]. Fully activated HSCs release and upregulate expression of TIMP-1 and TIMP-2, which inactivate MMPs through proteolytic cleavage [43][45][48]. Targeting activated HSCs in vivo decreased the expression of TIMP-1 and TIMP-2 and resulted in attenuated liver fibrosis [49]. Impairment of HSCs activation in mice downregulated TIMP-1 and diminished alcohol-induced steatohepatitis [50]. Interestingly, the addition of activated MMP-2 to aHSCs in culture enhanced the apoptosis of HSCs [51].
Increased production of ECM proteins, such as collagen I and laminin-5 (Ln-5), is associated with the growth and metastasis of HCC. Collagen I promotes HCC cell proliferation by regulating the integrin β1/FAK signaling pathway [52]. HSCs produce collagen I [53], which has been associated with the increased aggressiveness of HCC [54], where silencing its expression in HSCs may treat liver fibrosis [55].
Upregulation of Ln-5 in HCC patients promotes the migration of tumor cells, which is directly related to poor prognosis and tumor metastasis [56]. HCC grows in a microenvironment enriched with Ln-5 produced by HSCs [57]. In human HCC tissues, Ln-5 was distributed mainly along aHSCs, stimulating tumor cell migration [58]. Blocking antibodies against Ln-5 in HCC cell lines in the presence of HSCs inhibited tumor metastasis [58], while the presence of HSCs or Ln-5 in HCC cell lines increased resistance to sorafenib [57].
Normalization of ECM may represent an important therapeutic strategy for HCC [59]. Analysis of HSC-secreted proteins that control components of ECM could identify possible targets for HCC treatment.
This entry is adapted from the peer-reviewed paper 10.3390/ijms232315292
References
- Ramani, A.; Tapper, E.B.; Griffin, C.; Shankar, N.; Parikh, N.D.; Asrani, S.K. Hepatocellular Carcinoma-Related Mortality in the USA, 1999–2018. Am. J. Dig. Dis. 2022, 67, 4100–4111.
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. 2021, 149, 1–61. Adv. Cancer Res.
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 153–186.
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522.
- Kanel, G.C.; Korula, J. General Aspects of the Liver and Liver Diseases. Atlas Liver Pathol. 2011, 3–15.
- Barry, A.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709.
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363.
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer Find the latest version: Review series Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 2013, 123, 1902–1910.
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667.
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843.
- Mossenta, M.; Busato, D.; Dal Bo, M.; Macor, M.; Toffoli, G. Novel Nanotechnology Approaches to Overcome Drug Resistance in the Treatment of Hepatocellular Carcinoma: Glypican 3 as a Useful Target for Innovative Therapies. Int. J. Mol. Sci. 2022, 23, 10038.
- Sarveazad, A.; Agah, S.; Babahajian, A.; Amini, N.; Bahardoust, M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J. Res. Med Sci. Off. J. Isfahan Univ. Med. Sci. 2019, 24, 86.
- Hemminki, K.; Försti, A.; Hemminki, O.; Liska, V.; Hemminki, A. Long-term survival trends for primary liver and pancreatic cancers in the Nordic countries. JHEP Reports 2022, 4, 100602.
- Ruan, Q.; Wang, H.; Burke, L.J.; Bridle, K.R.; Li, X.; Zhao, C.-X.; Crawford, D.H.G.; Roberts, M.; Liang, X. Therapeutic modulators of hepatic stellate cells for hepatocellular carcinoma. Int. J. Cancer 2020, 147, 1519–1527.
- Wu, M.; Miao, H.; Fu, R.; Zhang, J.; Zheng, W. Hepatic Stellate Cell: A Potential Target for Hepatocellular Carcinoma. Curr. Mol. Pharmacol. 2020, 13, 261–272.
- Zhao, W.; Zhang, L.; Yin, Z.; Su, W.; Ren, G.; Zhou, C.; You, J.; Fan, J.; Wang, X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int. J. Cancer 2011, 129, 2651–2661.
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668.
- Zheng, X.; Jin, W.; Wang, S.; Ding, H. Progression on the Roles and Mechanisms of Tumor-Infiltrating T Lymphocytes in Patients With Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 1480.
- Xu, Y.; Huang, Y.; Xu, W.; Zheng, X.; Yi, X.; Huang, L.; Wang, Y.; Wu, K. Activated Hepatic Stellate Cells (HSCs) Exert Immunosuppressive Effects in Hepatocellular Carcinoma by Producing Complement C3. OncoTargets Ther. 2020, ume 13, 1497–1505.
- Charles, R.; Chou, H.-S.; Wang, L.; Fung, J.; Lu, L.; Qian, S. Human Hepatic Stellate Cells Inhibit T-Cell Response Through B7-H1 Pathway. Transplantation 2013, 96, 17–24.
- Schildberg, F.A.; Wojtalla, A.; Siegmund, S.V.; Endl, E.; Diehl, L.; Abdullah, Z.; Kurts, C.; Knolle, P.A. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology 2011, 54, 262–272.
- Zhao, W.; Su, W.; Kuang, P.; Zhang, L.; Liu, J.; Yin, Z.; Wang, X. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. Int. J. Oncol. 2012, 41, 457–464.
- Li, X.; Su, Y.; Hua, X.; Xie, C.; Liu, J.; Huang, Y.; Zhou, L.; Zhang, M.; Li, X.; Gao, Z. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J. Transl. Med. 2017, 15, 1–11.
- Cheng, J.-N.; Yuan, Y.-X.; Zhu, B.; Jia, Q. Myeloid-Derived Suppressor Cells: A Multifaceted Accomplice in Tumor Progression. Front. Cell Dev. Biol. 2021, 9.
- Xu, Y. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget 2016, 7, 8866–8878.
- Lee, H.L.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Han, N.I.; Yoon, S.K. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 2019, 9, 3260.
- Li, J.; Lau, G.K.-K.; Chen, P.L.; Dong, S.-S.; Lan, H.Y.; Huang, X.-R.; Li, Y.; Luk, J.; Yuan, Y.; Guan, X.-Y. Interleukin 17A Promotes Hepatocellular Carcinoma Metastasis via NF-kB Induced Matrix Metalloproteinases 2 and 9 Expression. PLoS ONE 2011, 6, e21816.
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.-X.; He, H.-W.; Cai, X.-Y.; Zhou, J.; Cheng, Y.-F.; Fan, J.; et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3–11.
- Ichikawa, S.; Mucida, D.; Tyznik, A.J.; Kronenberg, M.; Cheroutre, H. Hepatic Stellate Cells Function as Regulatory Bystanders. J. Immunol. 2011, 186, 5549–5555.
- Ricketts, T.D.; Prieto-Dominguez, N.; Gowda, P.S.; Ubil, E. Mechanisms of Macrophage Plasticity in the Tumor Environment: Manipulating Activation State to Improve Outcomes. Front. Immunol. 2021, 12.
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602.
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900.
- Liu, G.; Yin, L.; Ouyang, X.; Zeng, K.; Xiao, Y.; Li, Y. M2 Macrophages Promote HCC Cells Invasion and Migration via miR-149-5p/MMP9 Signaling. J. Cancer 2020, 11, 1277–1287.
- Yeung, O.W.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616.
- Xi, S.; Zheng, X.; Li, X.; Jiang, Y.; Wu, Y.; Gong, J.; Jie, Y.; Li, Z.; Cao, J.; Sha, L.; et al. Activated Hepatic Stellate Cells Induce Infiltration and Formation of CD163+ Macrophages via CCL2/CCR2 Pathway. Front. Med. 2021, 8, 627927.
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 5381.
- Yu, M.; Li, Z. Natural killer cells in hepatocellular carcinoma: Current status and perspectives for future immunotherapeutic approaches. Front. Med. 2017, 11, 509–521.
- Mossanen, J.C.; Tacke, F. Role of lymphocytes in liver cancer. OncoImmunology 2013, 2, e26468.
- Glässner, A.; Eisenhardt, M.; Krämer, B.; Körner, C.; Coenen, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Investig. 2012, 92, 967–977.
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-Dependent and Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Dependent Manners. Gastroenterology 2006, 130, 435–452.
- Xia, T.-H. Tumor-specific hepatic stellate cells (tHSCs) induces DIgR2 expression in dendritic cells to inhibit T cells. Oncotarget 2017, 8, 55084–55093. Available online: www.impactjournals.com/oncotarget/ (accessed on 15 August 2017).
- HCC Monitor. New evidence supports a key role of the immune system in HCC, HCC monitor. Target. Oncol. 2016, 2, 3. Available online: https://www.targetedonc.com (accessed on 15 August 2017).
- Zois, C.D.; Baltayiannis, G.H.; Karayiannis, P.; Tsianos, E.V. Systematic review: Hepatic fibrosis-regression with therapy. Aliment. Pharmacol. Ther. 2008, 28, 1175–1187.
- Arriazu, E.; de Galarreta, M.R.; Cubero, F.J.; Varela-Rey, M.; de Obanos, M.P.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular Matrix and Liver Disease. Antioxidants Redox Signal. 2014, 21, 1078–1097.
- Jung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228.
- Sun, M.; Kisseleva, Y. Reversibility of liver fibrosis. In Clinics and Research in Hepatology and Gastroenterology; Elsevier Masson SAS: Amsterdam, The Netherlands, 2015; pp. S60–S63.
- Kisseleva, T.; Brenner, D. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2006, 21, S84–S87.
- Arthur, M.J.; Mann, D.A.; Iredale, J.P. Tissue inhibitors of metalloproteinases, hepatic stellate cells and liver fibrosis. J. Gastroenterol. Hepatol. 1998, 13, S33–S38.
- Zhang, Y.; Li, Y.; Mu, T.; Tong, N.; Cheng, P. Hepatic stellate cells specific liposomes with the Toll-like receptor 4 shRNA attenuates liver fibrosis. J. Cell. Mol. Med. 2021, 25, 1299–1313.
- Arab, J.P.; Cabrera, D.; Sehrawat, T.S.; Jalan-Sakrikar, N.; Verma, V.K.; Simonetto, D.; Cao, S.; Yaqoob, U.; Leon, J.; Freire, M.; et al. Hepatic stellate cell activation promotes alcohol-induced steatohepatitis through Igfbp3 and SerpinA12. J. Hepatol. 2020, 73, 149–160.
- Hartland, S.N.; Murphy, F.; Aucott, R.L.; Abergel, A.; Zhou, X.; Waung, J.; Patel, N.; Bradshaw, C.; Collins, J.; Mann, D.; et al. Active matrix metalloproteinase-2 promotes apoptosis of hepatic stellate cells via the cleavage of cellular N-cadherin. Liver Int. 2009, 29, 966–978.
- Zheng, X.; Liu, W.; Xiang, J.; Liu, P.; Ke, M.; Wang, B.; Lv, Y. Collagen I promotes hepatocellular carcinoma cell proliferation by regulating integrin β1/FAK signaling pathway in nonalcoholic fatty liver. Oncotarget 2017, 8, 95586–95595. Available online: www.impactjournals.com/oncotarget (accessed on 10 November 2017).
- Zhang, R.; Ma, M.; Lin, X.-H.; Liu, H.-H.; Chen, J.; Chen, J.; Gao, D.-M.; Cui, J.-F.; Ren, Z.-G.; Chen, R.-X. Extracellular matrix collagen I promotes the tumor progression of residual hepatocellular carcinoma after heat treatment. BMC Cancer 2018, 18, 901.
- Ma, H.-P.; Chang, H.-L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.-Y.; Wu, A.T.H.; Yeh, C.-T.; Tsai, S.-H.; Lee, W.-H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786.
- Yu, F.; Lin, Z.; Zheng, J.; Gao, S.; Lu, Z.; Dong, P. Suppression of collagen synthesis by Dicer gene silencing in hepatic stellate cells. Mol. Med. Rep. 2014, 9, 707–714.
- Giannelli, G.; Azzariti, A.; Fransvea, E.; Porcelli, L.; Antonaci, S.; Paradiso, A. Laminin-5 offsets the efficacy of gefitinib (‘Iressa’) in hepatocellular carcinoma cells. Br. J. Cancer 2004, 91, 1964–1969.
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Giannelli, G. Hepatic Stellate Cells Induce Hepatocellular Carcinoma Cell Resistance to Sorafenib Through the Laminin-332/a3 Integrin Axis Recovery of Focal Adhesion Kinase Ubiquitination. Hepatology 2016, 64, 2103–2117.
- Santamato, A.; Fransvea, E.; Dituri, F.; Caligiuri, A.; Quaranta, M.; Niimi, T.; Pinzani, M.; Antonaci, S.; Giannelli, G. Hepatic stellate cells stimulate HCC cell migration via laminin-5 production. Clin. Sci. 2011, 121, 159–168.
- Wu, X.Z.; Chen, D.; Xie, G.R. Extracellular matrix remodeling in hepatocellular carcinoma: Effects of soil on seed? Med. Hypotheses 2006, 66, 1115–1120.
This entry is offline, you can click here to edit this entry!