The term hypercoordination refers to the extent of the coordination of an element by its normal value. In the hypercoordination sphere, the element can achieve planar and/or non-planar molecular shape. Hence, planar hypercoordinate carbon species violate two structural rules: (i) The highest coordination number of carbon is four and (ii) the tetrahedral orientation by the connected elements and/or groups. The unusual planar orientations are mostly stabilized by the electronic interactions of the central atom with the surrounding ligands. Primary knowledge of the planar hypercoordinate chemistry will lead to its forthcoming expansion. Experimental and theoretical interests in planar tetracoordinate carbon (ptC), planar pentacoordinate carbon (ppC), and planar hexacoordinate carbon (phC) are continued. The proposed electronic and mechanical strategies are helpful for the designing of the ptC compounds. Moreover, the 18-valence electron rule can guide the design of new ptC clusters computationally as well as experimentally. However, the counting of 18-valence electrons is not a requisite condition to contain a ptC in a cluster. Furthermore, this ptC idea is expanded to the probability of a greater coordination number of carbon in planar orientations. Unfortunately, until now, there are no such logical approaches to designing ppC, phC, or higher-coordinate carbon molecules/ions. There exist a few global minimum structures of phC clusters identified computationally, but none have been detected experimentally. All planar hypercoordinate carbon species in the global minima may be feasible in the gas phase.
- anti-van’t Hoff Le Bel
- planar tetracoordinate carbon
- planar pentacoordinate carbon
- planar hexacoordinate carbon
1. Planar Tetracoordinate Carbons (ptCs)
1.1. How to Achieve ptCs
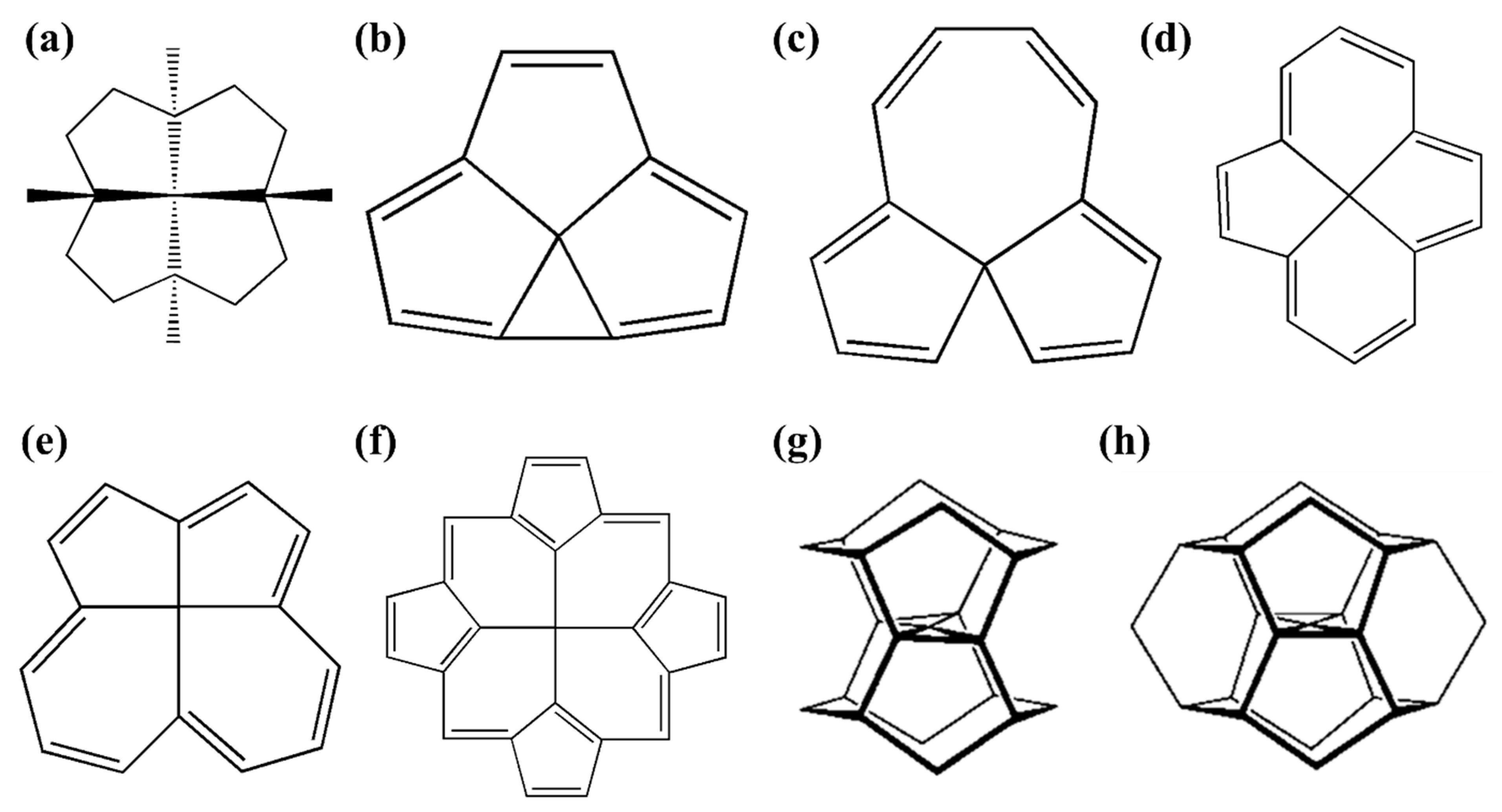
1.2. Early Examples of ptCs
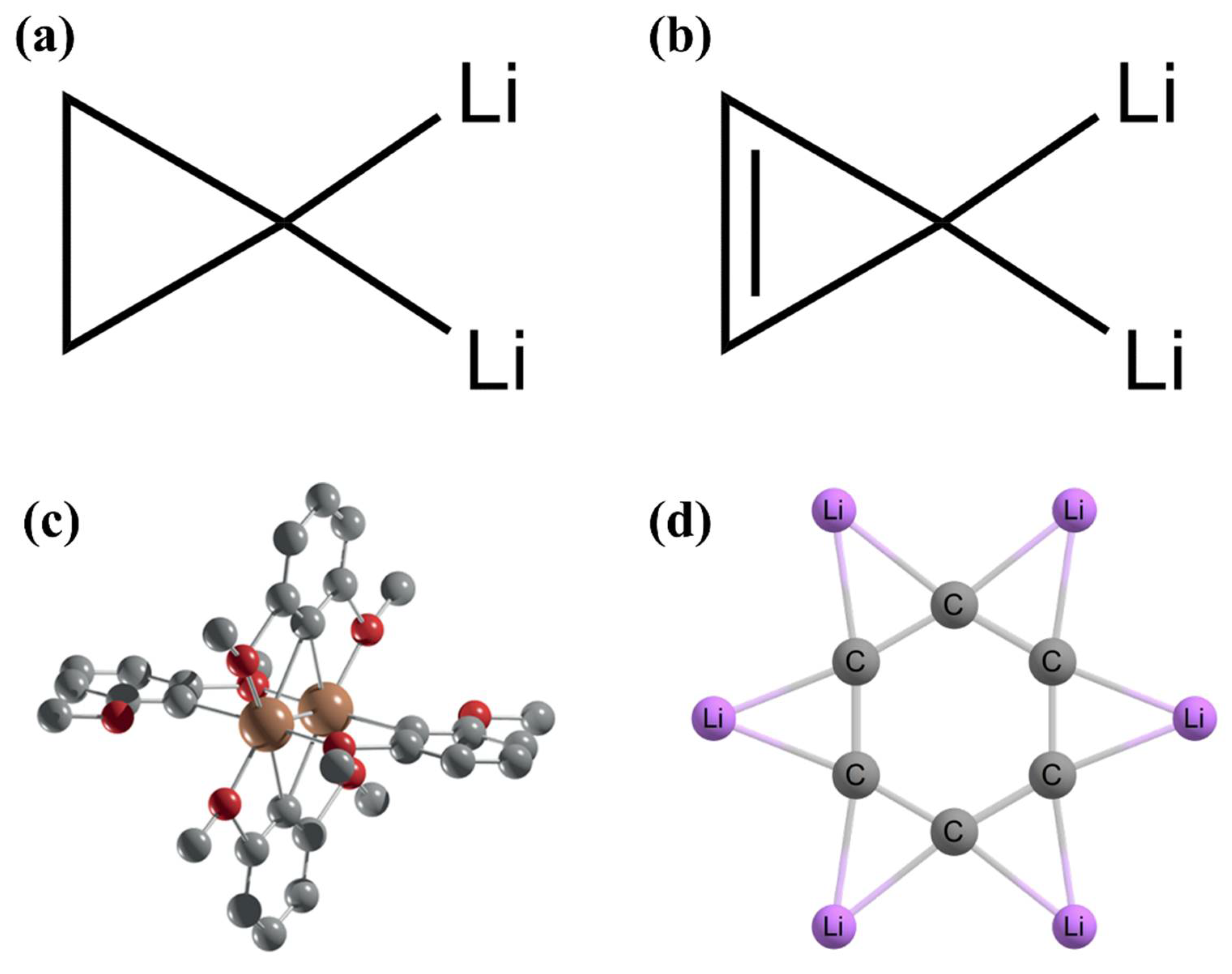
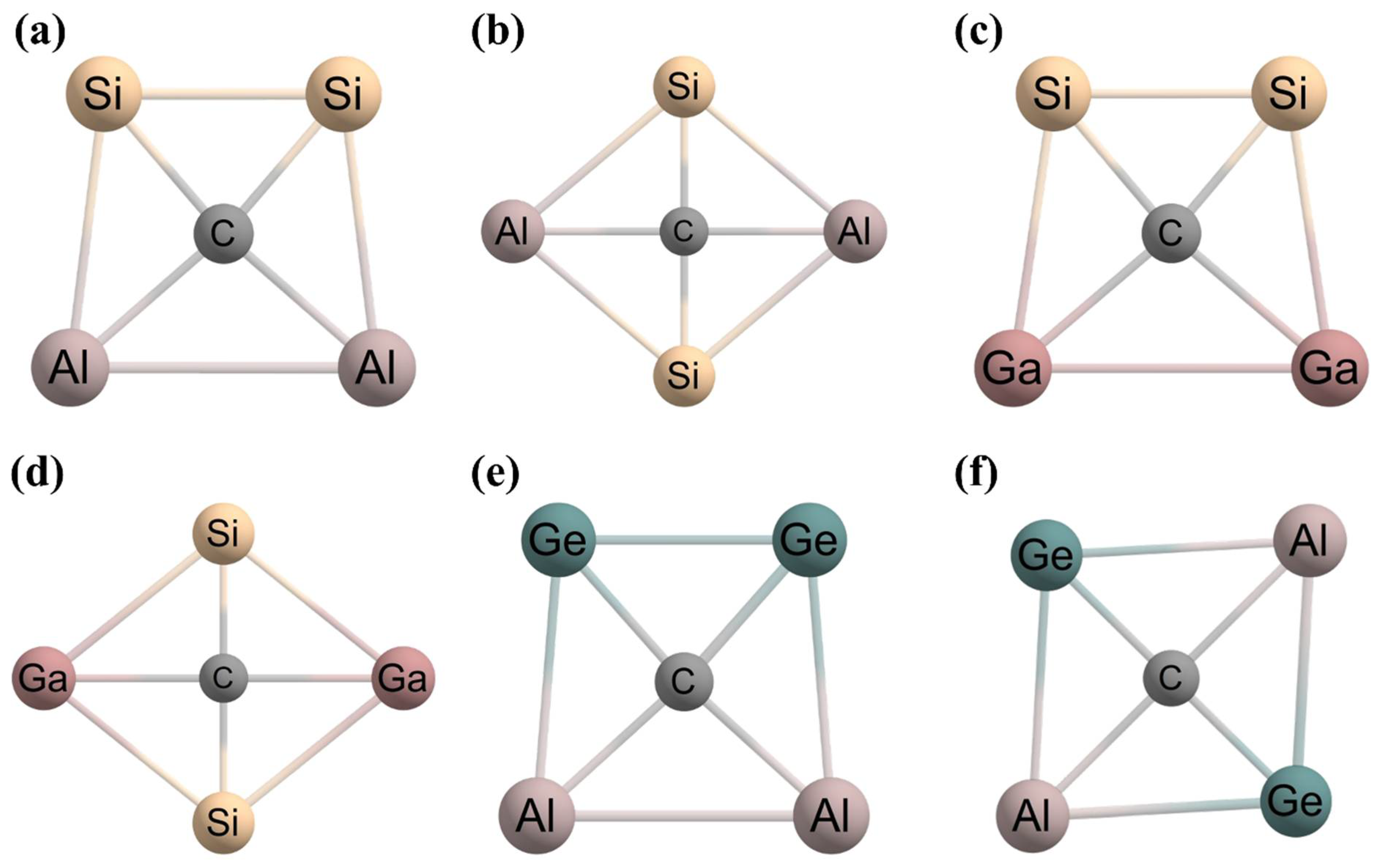
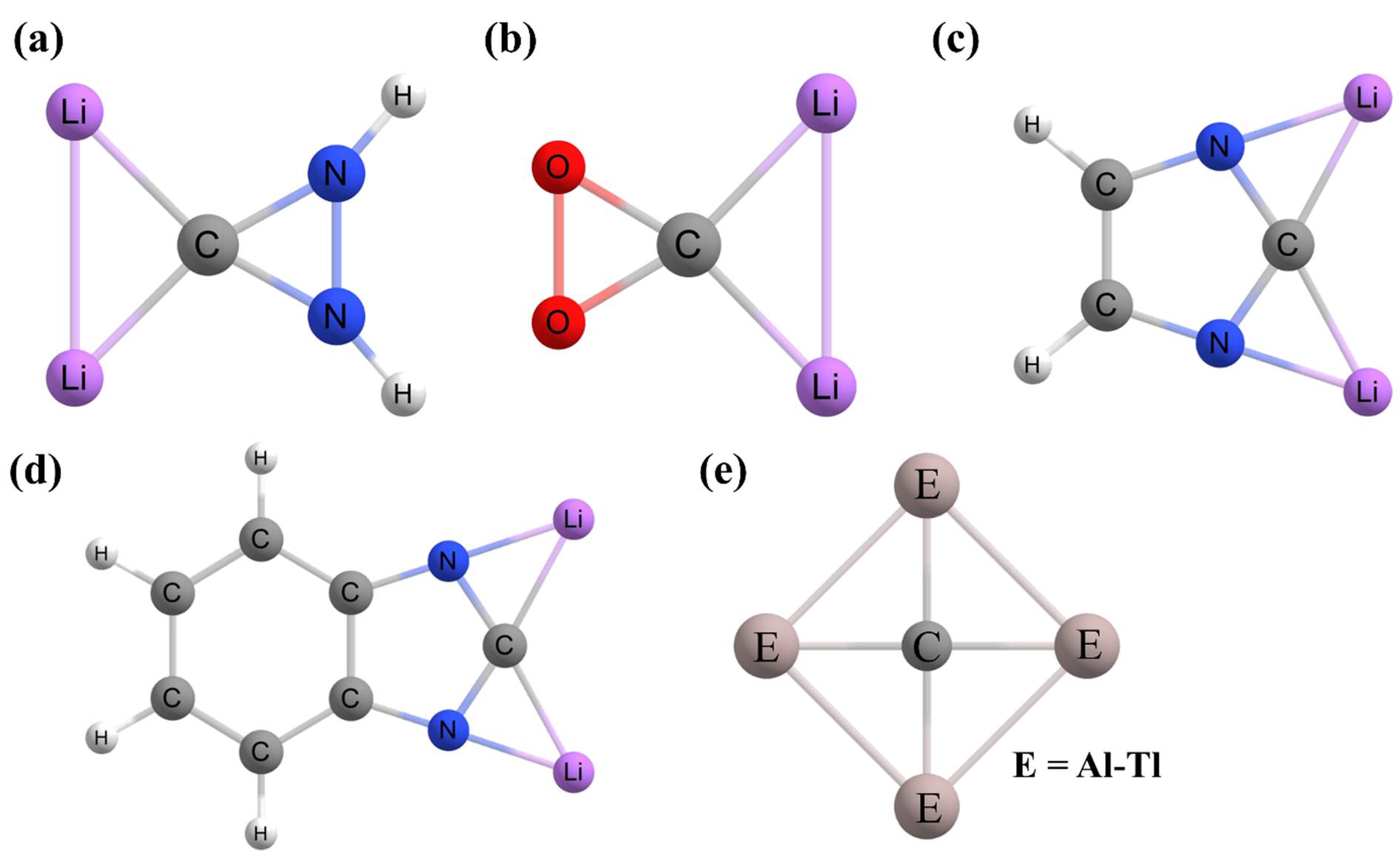
2. Planar Pentacoordinate Carbons (ppCs)
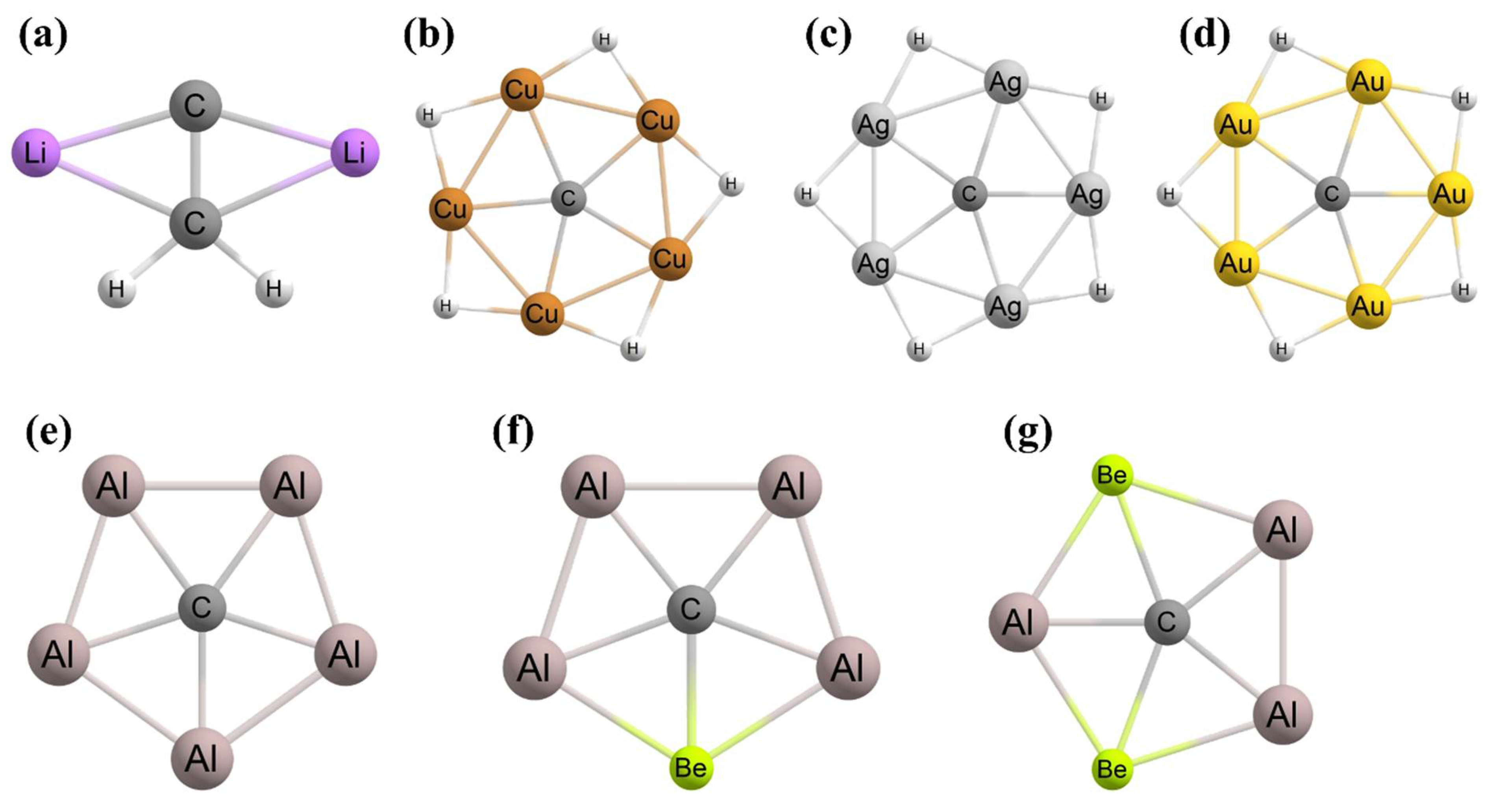
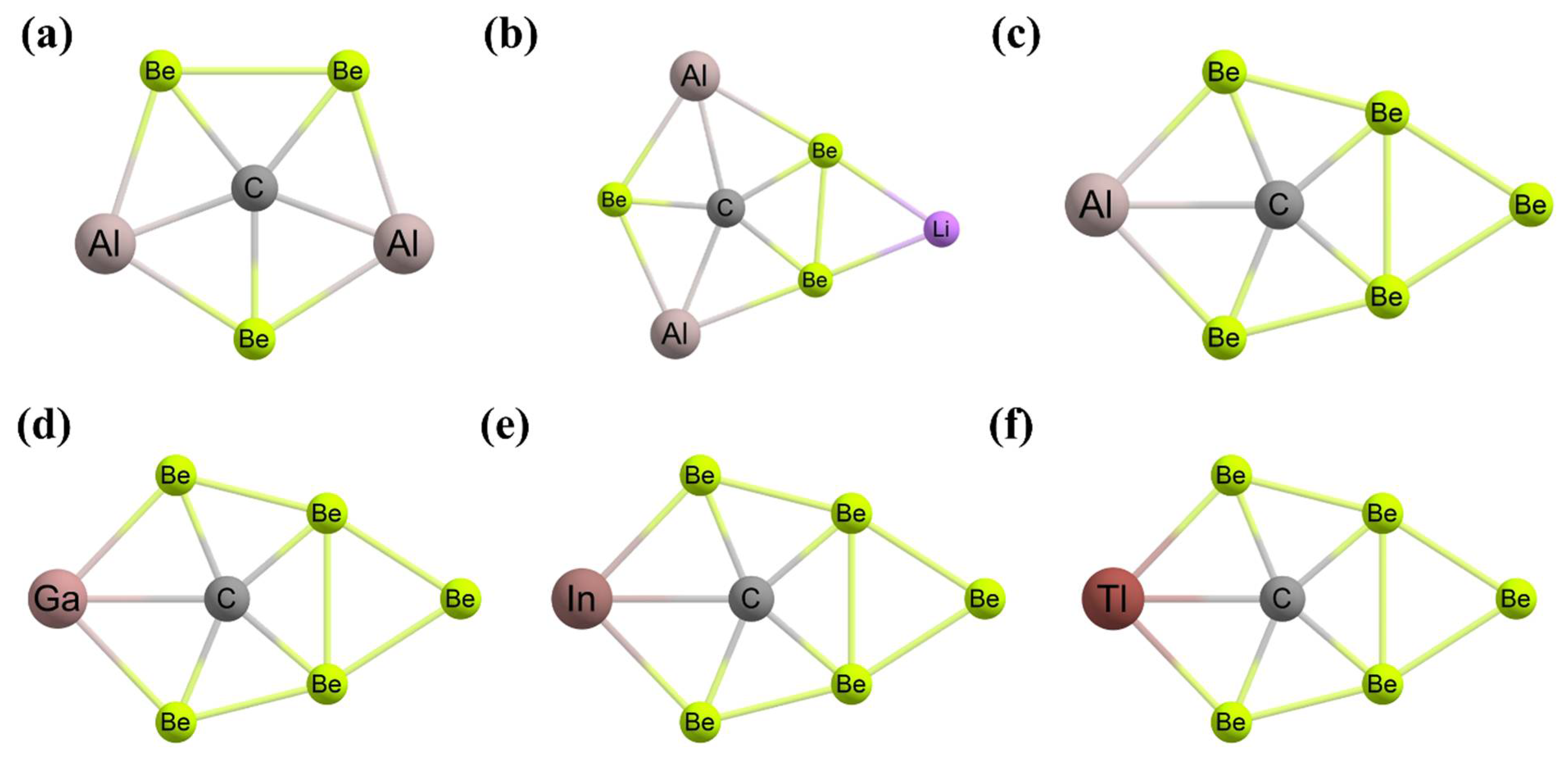
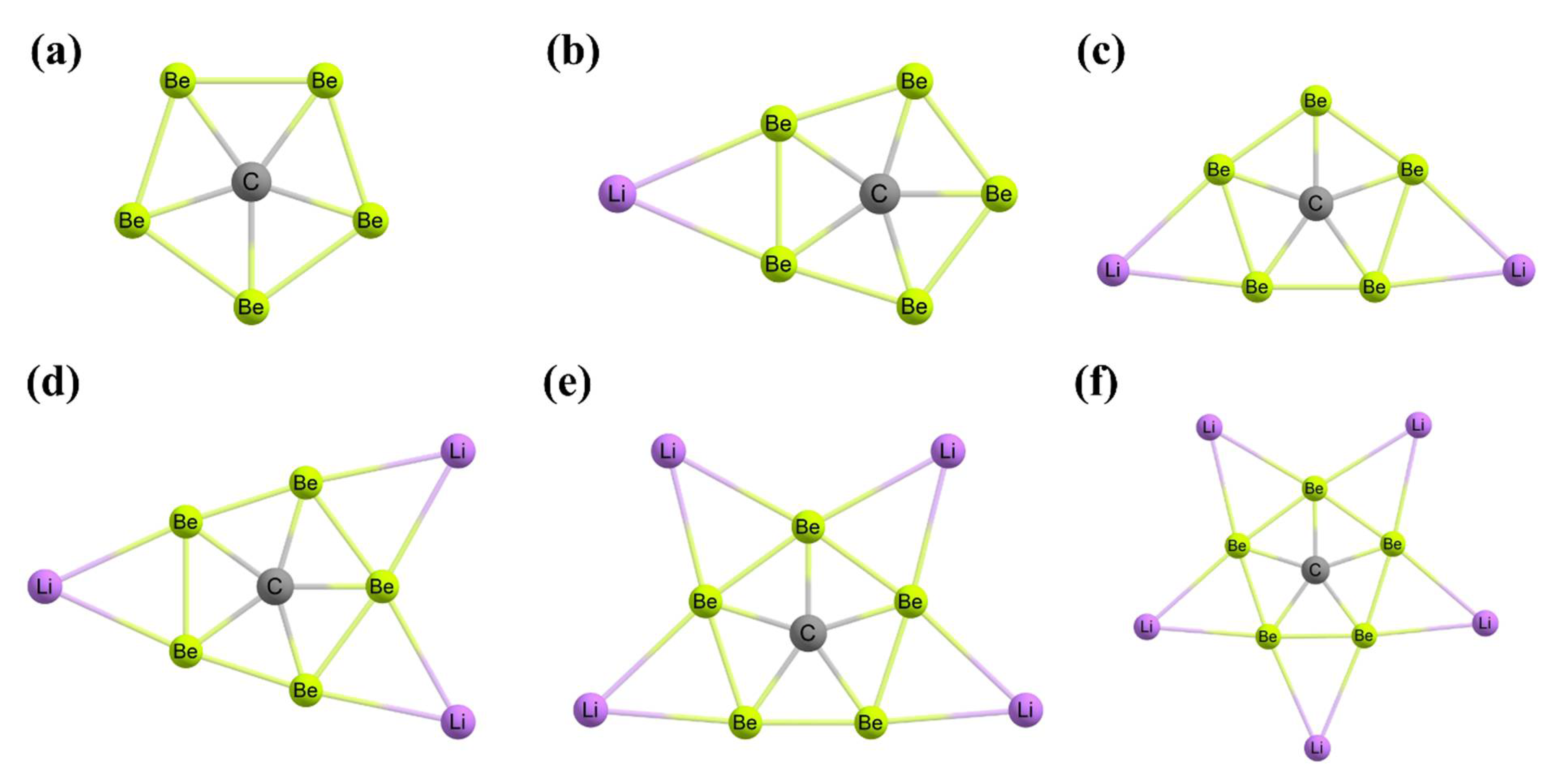
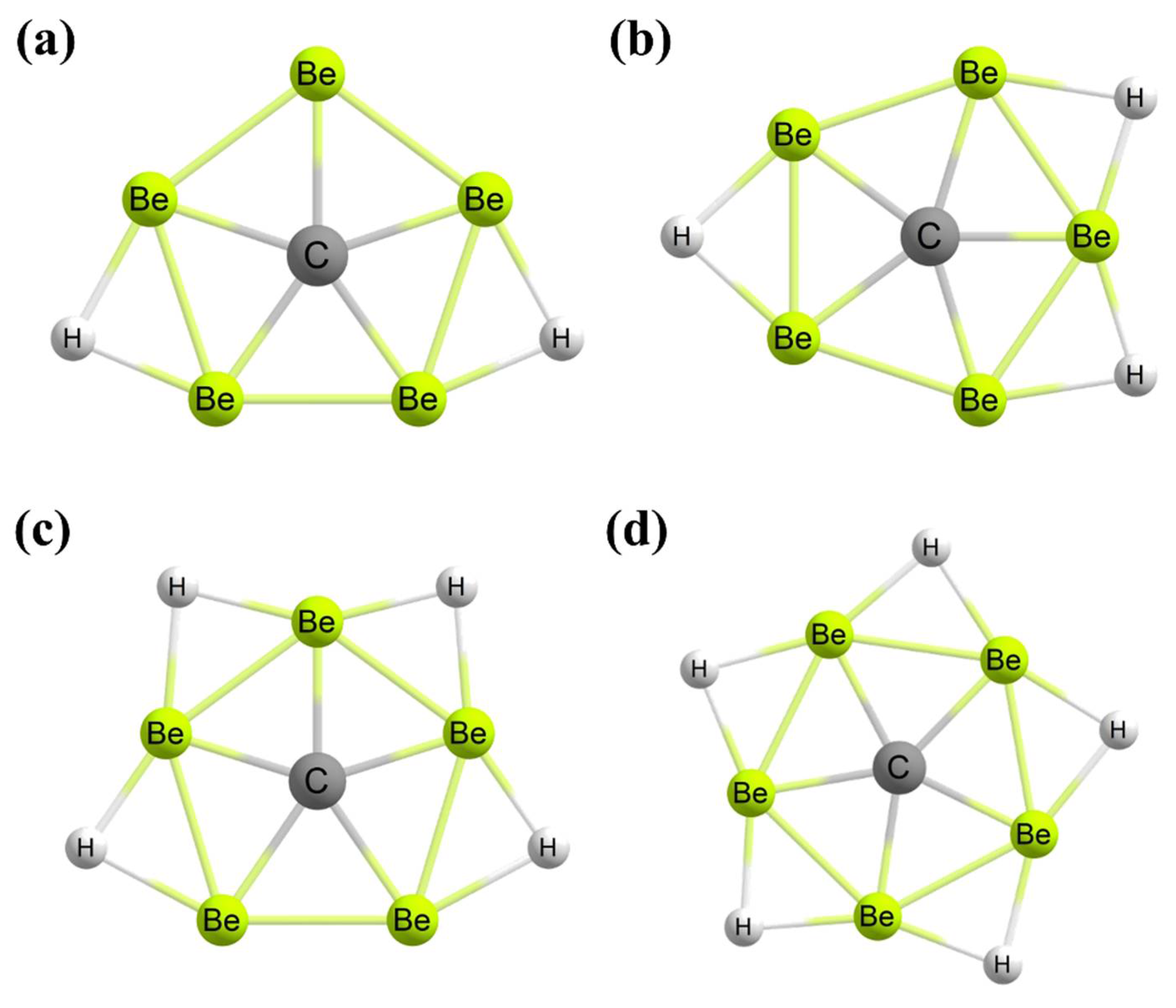
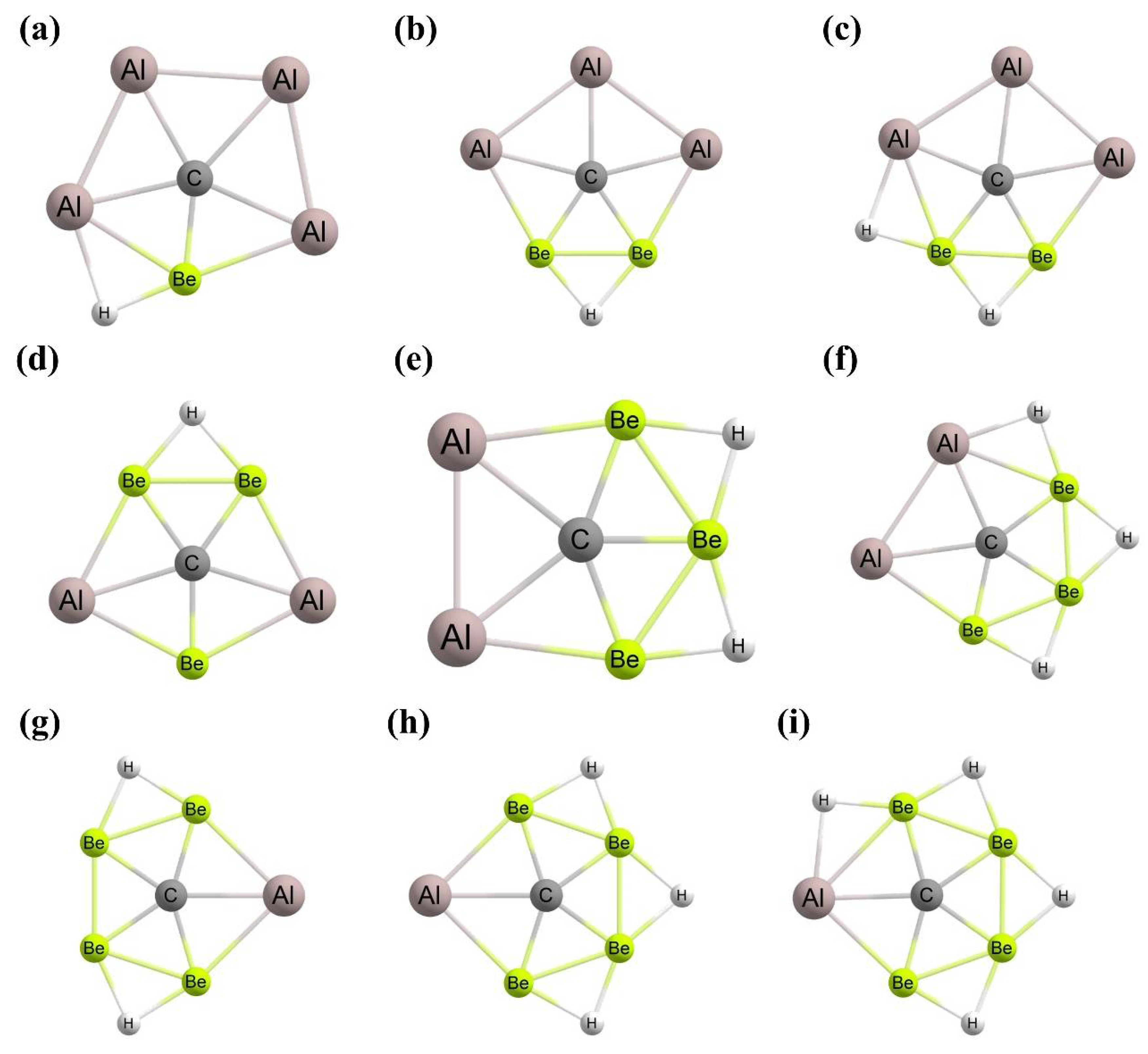
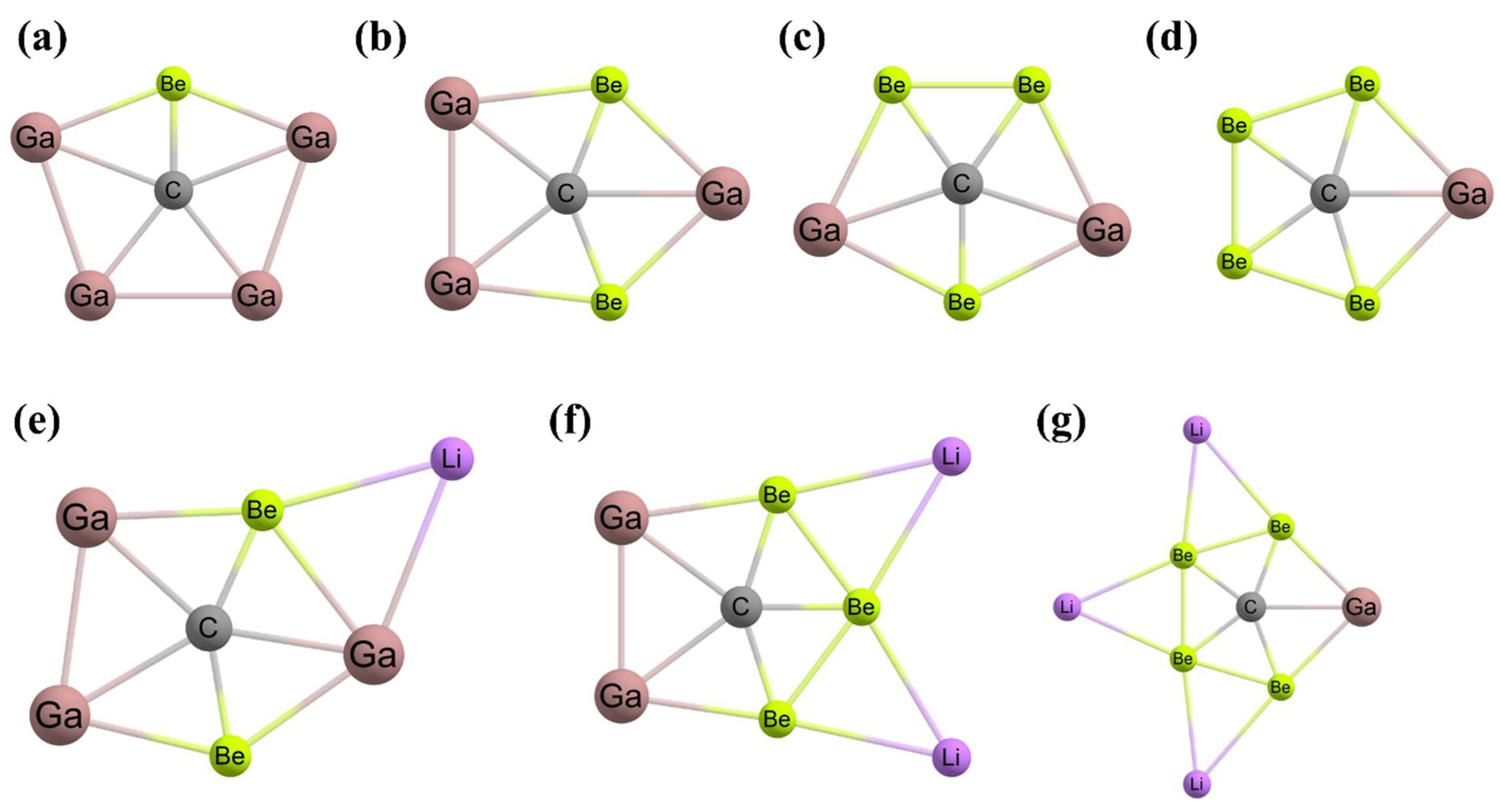
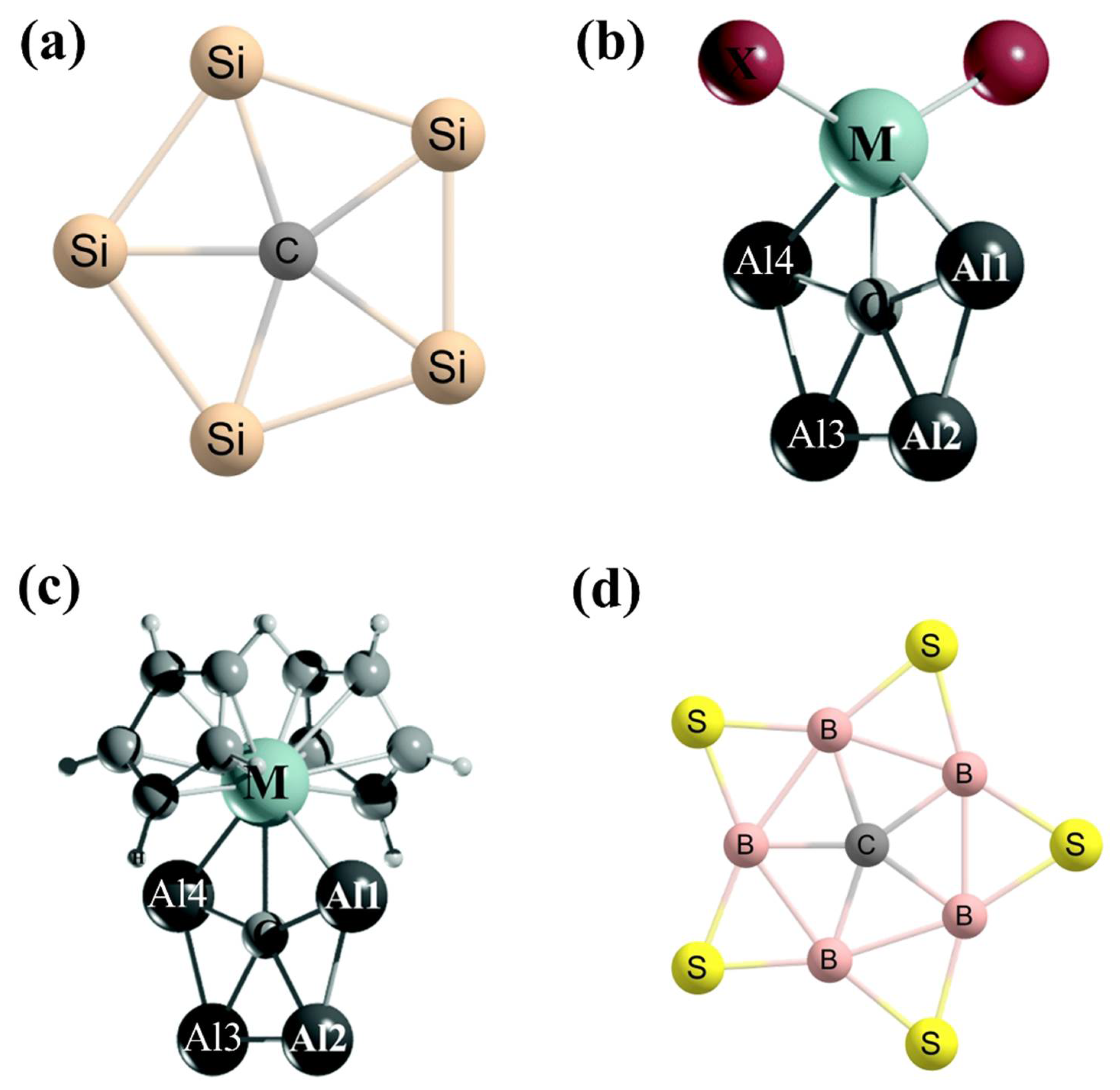
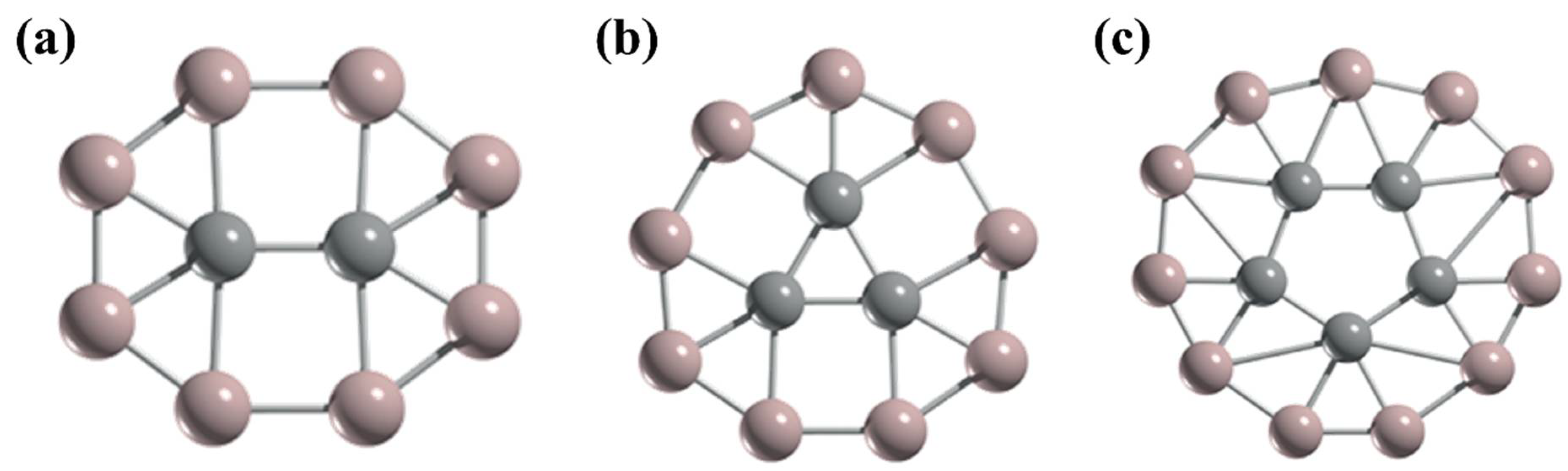
3. Planar Hexacoordinate Carbons (phCs)

This entry is adapted from the peer-reviewed paper 10.3390/chemistry4040113
References
- Pepper, M.J.; Shavitt, I.; Schleyer, P.v.R.; Glukhovtsev, M.N.; Janoschek, R.; Quack, M. Is the stereomutation of methane possible? J. Comput. Chem. 1995, 16, 207–225.
- Golden, D.M.; Walsh, R.; Benson, S.W. The Thermochemistry of the Gas Phase Equilibrium I2 + CH4 CH3I + HI and the Heat of Formation of the Methyl Radical. J. Am. Chem. Soc. 1965, 87, 4053–4057.
- Crans, D.C.; Snyder, J.P. Tetracoordinate planar carbon: A singlet biradical. J. Am. Chem. Soc. 1980, 102, 7152–7154.
- Schleyer, P.v.R.; Boldyrev, A.I. A new, general strategy for achieving planar tetracoordinate geometries for carbon and other second row periodic elements. J. Chem. Soc. Chem. Commun. 1991, 1536–1538.
- Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in Pentaatomic Molecules. J. Am. Chem. Soc. 1998, 120, 7967–7972.
- Gribanova, T.N.; Minyaev, R.M.; Minkin, V.I. Planar Tetracoordinate Carbon in Organoboron Compounds: Ab initio Computational Study. Collect. Czech. Chem. Commun. 1999, 64, 1780–1789.
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and ab Initio Study. J. Am. Chem. Soc. 1999, 121, 6033–6038.
- Wang, Z.X.; Manojkumar, T.K.; Wannere, C.; Schleyer, P.V.R. A Theoretical Prediction of Potentially Observable Lithium Compounds with Planar Tetracoordinate Carbons. Org. Lett. 2001, 3, 1249–1252.
- Wang, Z.X.; Schleyer, P.v.R. A New Strategy To Achieve Perfectly Planar Carbon Tetracoordination. J. Am. Chem. Soc. 2001, 123, 994–995.
- Merino, G.; Méndez-Rojas, M.A.; Vela, A. (C5M2-n)n- (M = Li, Na, K, and n = 0, 1, 2). A New Family of Molecules Containing Planar Tetracoordinate Carbons. J. Am. Chem. Soc. 2003, 125, 6026–6027.
- Sahin, Y.; Prasang, C.; Hofmann, M.; Subramanian, G.; Geiseler, G.; Massa, W.; Berndt, A. A Diboracyclopropane with a Planar-Tetracoordinate Carbon Atom and a Triborabicyclobutane. Angew. Chem. Int. Ed. 2003, 42, 671–674.
- Li, S.D.; Ren, G.M.; Miao, C.Q.; Jin, Z.H. M4H4X: Hydrometals (M=Cu, Ni) Containing Tetracoordinate Planar Nonmetals (X = B, C, N, O). Angew. Chem. Int. Ed. 2004, 43, 1371–1373.
- Merino, G.; Méndez-Rojas, M.A.; Beltran, H.I.; Corminboeuf, C.; Heine, T.; Vela, A. Theoretical Analysis of the Smallest Carbon Cluster Containing a Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 2004, 126, 16160–16169.
- Pancharatna, P.D.; Méndez-Rojas, M.A.; Merino, G.; Vela, A.; Hoffmann, R. Planar Tetracoordinate Carbon in Extended Systems. J. Am. Chem. Soc. 2004, 126, 15309–15315.
- Priyakumar, U.D.; Reddy, A.S.; Sastry, G.N. The design of molecules containing planar tetracoordinate carbon. Tetrahedron Lett. 2004, 45, 2495–2498.
- Merino, G.; Méndez-Rojas, M.A.; Vela, A.; Heine, T. Recent advances in planar tetracoordinate carbon chemistry. J. Comput. Chem. 2007, 28, 362–372.
- Minkin, V.I.; Gribanova, T.N.; Minkin, V.I.; Starikov, A.G.; Hoffmann, R. Planar and Pyramidal Tetracoordinate Carbon in Organoboron Compounds. J. Org. Chem. 2005, 70, 6693–6704.
- Su, M.D. Theoretical Designs for Planar Tetracoordinated Carbon in Cu, Ag, and Au Organometallic Chemistry: A New Target for Synthesis. Inorg. Chem. 2005, 44, 4829–4833.
- Li, S.D.; Ren, G.M.; Miao, C.Q. (M4H3X)2B2O2: Hydrometal Complexes (M = Ni, Mg) Containing Double Tetracoordinate Planar Nonmetal Centers (X = C, N). J. Phys. Chem. A 2005, 109, 259–261.
- Esteves, P.M.; Ferreira, N.B.P.; Corrêa, R.J. Neutral Structures with a Planar Tetracoordinated Carbon Based on Spiropentadiene Analogues. J. Am. Chem. Soc. 2005, 127, 8680–8685.
- Erker, G. Stereochemistry and catalysis with zirconium complexes. Pure Appl. Chem. 1991, 63, 797–806.
- Erker, G.; Albrecht, M.; Kruger, C.; Werner, S. Novel synthetic route to hydrocarbyl-bridged dinuclear zirconium/aluminum complexes exhibiting a planar tetracoordinate carbon center. Organometallics 1991, 10, 3791–3793.
- Albrecht, M.; Erker, G.; Nolte, M.; Kruger, C. Planar tetracoordinate carbon stabilized in a dimetallic hafnium/aluminium compound: Formation and crystal structure of Cp2HfA1Me2. J. Organomet. Chem. 1992, 427, C21–C25.
- Rottger, D.; Erker, G.; Frohlich, R.; Grehl, M.; Silverio, S.J.; Hylakryspin, I.; Gleiter, R. Determination of the Stabilization Energy of Planar-Tetracoordinate Carbon in Dynamic Dinuclear (μ-Hydrocarbyl)bis(zirconocene) Cation Complexes and Detection of an Organometallic Memory Effect in Their Formation. J. Am. Chem. Soc. 1995, 117, 10503–10512.
- Rottger, D.; Erker, G.; Frohlich, R. Formation of stable organometallic planar-tetracoordinate carbon compounds containing a cationic (μ-R1CCR2) framework. J. Organomet. Chem. 1996, 518, 221–225.
- Rottger, D.; Erker, G.; Frohlich, R.; Kotila, S. Stabilization of a Planar-tetracoordinate Carbon Center in an Organometallic Complex Containing Both a Zirconocene and a Hafnocene Moiety. Chem. Ber. 1996, 129, 1–3.
- Schottek, J.; Erker, G.; Frohlich, R. Formation of Metallocene-Stabilized Planar-Tetracoordinate Carbon Compounds by a Protonation Route. Eur. J. Inorg. Chem. 1998, 1998, 551–558.
- Choukroun, R.; Donnadieu, B.; Zhao, J.S.; Cassoux, P.; Lepetit, C.; Silvi, B. Synthesis and Characterization of Heterodimetallic Complexes (Cp’ = C5H4t-Bu, C5H4Me). Formation Mechanism and Theoretical (ELF) Evidence for the Existence of Planar Tetracoordinate Carbon (ptC). Organometallics 2000, 19, 1901–1911.
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.D.; Boldyrev, A.I. Pentaatomic Tetracoordinate Planar Carbon, 2−: A New Structural Unit and Its Salt Complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632.
- Hoffmann, R. The theoretical design of novel stabilized systems. Pure Appl. Chem. 1971, 28, 181–194.
- Sorger, K.; Schleyer, P.v.R. Planar and inherently non-tetrahedral tetracoordinate carbon: A status report. J. Mol. Struct. THEOCHEM 1995, 338, 317–346.
- Röttger, D.; Erker, G. Compounds containing planar-tetracoordinate carbon. Angew. Chem. Int. Ed. 1997, 36, 812–827.
- Radom, L.; Rasmussen, D.R. The planar carbon story. Pure Appl. Chem. 1998, 70, 1977–1984.
- Erker, G. Using bent metallocenes for stabilizing unusual coordination geometries at carbon. Chem. Soc. Rev. 1999, 28, 307–314.
- Siebert, W.; Gunale, A. Compounds containing a planar-tetracoordinate carbon atom as analogues of planar methane. Chem. Soc. Rev. 1999, 28, 367–371.
- Keese, R. Carbon flatland: planar tetracoordinate carbon and fenestrenes. Chem. Rev. 2006, 106, 4787–4808.
- McGrath, M.P.; Radom, L. Alkaplanes: A class of neutral hydrocarbons containing a potentially planar tetracoordinate carbon. J. Am. Chem. Soc. 1993, 115, 3320–3321.
- Lyons, J.E.; Rasmussen, D.R.; McGrath, M.P.; Nobes, R.H.; Radom, L. Octaplan: Ein gesättigter Kohlenwasserstoff mit ungewöhnlich niedriger Ionisierungsenergie und einem planar-tetrakoordinierten Kohlenstoffatom im Radikalkation. Angew. Chem. Int. Ed. Engl. 1994, 33, 1667–1668.
- Ding, B.W.; Keese, R.; Stoeckli-Evans, H. First Synthesis and Structure of a Tetraazasilafenestrane. Angew. Chem. Int. Ed. 1999, 38, 375–376.
- Rasmussen, D.R.; Radom, L. Planar tetrakoordinierter Kohlenstoff in einem neutralen gesättigten Kohlenwasserstoff: Theoretischer Entwurf und Charakterisierung. Angew. Chem. Int. Ed. 1999, 38, 2875–2878.
- Wang, Z.X.; Schleyer, P.v.R. The Theoretical Design of Neutral Planar Tetracoordinate Carbon Molecules with C(C)4 Substructures. J. Am. Chem. Soc. 2002, 124, 11979–11982.
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; Schleyer, P.v.R.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427.
- Cotton, F.A.; Millar, M. The probable existence of a triple bond between two vanadium atoms. J. Am. Chem. Soc. 1977, 99, 7886–7891.
- Xie, Y.; Schaefer, H.F. Hexalithiobenzene: A D6h equilibrium geometry with six lithium atoms in bridging positions. Chem. Phys. Lett. 1991, 179, 563–567.
- Wang, L.S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687.
- Vassilev-Galindo, V.; Pan, S.; Donald, K.J.; Merino, G. Planar pentacoordinate carbons. Nat. Rev. Chem. 2018, 2, 0114.
- Bolton, E.E.; Laidig, W.D.; Schleyer, P.v.R.; Schaefer, H.F. Does singlet 1,1-dilithioethene really prefer a perpendicular structure? J. Phys. Chem. 1995, 99, 17551–17557.
- Tsipis, C.A.; Karagiannis, E.E.; Kladou, P.F.; Tsipis, A.C. Aromatic Gold and Silver ‘Rings’: Hydrosilver(I) and Hydrogold(I) Analogues of Aromatic Hydrocarbons. J. Am. Chem. Soc. 2004, 126, 12916–12929.
- Tsipis, A.C.; Tsipis, C.A. Hydrometal Analogues of Aromatic Hydrocarbons: A New Class of Cyclic Hydrocoppers(I). J. Am. Chem. Soc. 2003, 125, 1136–1137.
- Li, S.D.; Miao, C.Q.; Ren, G.M. D5h Cu5H5X: Pentagonal hydrocopper Cu5H5 containing pentacoordinate planar nonmetal centers (X = B, C, N, O). Eur. J. Inorg. Chem. 2004, 2004, 2232–2234.
- Li, S.D.; Guo, Q.L.; Miao, C.Q.; Ren, G.M. Investigation on transition-metal hydrometal complexes MnHnC with planar coordinate carbon centers by density functional theory. Acta Phys. Chim. Sin. 2007, 23, 743–745.
- Pei, Y.; An, W.; Ito, K.; Schleyer, P.v.R.; Zeng, X.C. Planar pentacoordinate carbon in CAl5+: A global minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400.
- Jimenez-Halla, J.O.C.; Wu, Y.B.; Wang, Z.X.; Islas, R.; Heine, T.; Merino, G. CAl4Be and CAl3Be2−: Global Minima with a Planar Pentacoordinate Carbon Atom. Chem. Commun. 2010, 46, 8776–8778.
- Wu, Y.B.; Duan, Y.; Lu, H.G.; Li, S.D. CAl2Be32– and its salt complex LiCAl2Be3–: Anionic global minimawith planar pentacoordinate carbon. J. Phys. Chem. A 2012, 116, 3290–3294.
- Castro, A.C.; Martínez-Guajardo, G.; Johnson, T.; Ugalde, J.M.; Wu, Y.B.; Mercero, J.M.; Heine, T.; Donald, K.J.; Merino, G. CBe5E– (E = Al, Ga, In, Tl): Planar pentacoordinate carbon in heptaatomic clusters. Phys. Chem. Chem. Phys. 2012, 14, 14764–14768.
- Luo, Q. Theoretical observation of hexaatomic molecules containing pentacoordinate planar carbon. Sci. China, Ser. B Chem. 2008, 51, 1030–1035.
- Grande-Aztatzi, R.; Cabellos, J.L.; Islas, R.; Infante, I.; Mercero, J.M.; Restrepo, A.; Merino, G. Planar pentacoordinate carbons in CBe54– derivatives. Phys. Chem. Chem. Phys. 2015, 17, 4620–4624.
- Guo, J.C.; Ren, G.M.; Miao, C.Q.; Tian, W.J.; Wu, Y.B.; Wang, X. CBe5Hnn−4 (n = 2–5): Hydrogen-stabilized CBe5 pentagons containing planar or quasi-planar pentacoordinate carbons. J. Phys. Chem. A 2015, 119, 13101–13106.
- Guo, J.C.; Tian, W.J.; Wang, Y.J.; Zhao, X.F.; Wu, Y.B.; Zhai, H.J.; Li, S.D. Star-like superalkali cations featuring planar pentacoordinate carbon. J. Chem. Phys. 2016, 144, 244303.
- Zhao, X.F.; Bian, J.H.; Huang, F.; Yuan, C.; Wang, Q.; Liu, P.; Li, D.; Wang, X.; Wu, Y.B. Stabilization of beryllium-containing planar pentacoordinate carbon species through attaching hydrogen atoms. RSC Adv. 2018, 8, 36521–36526.
- Pan, S.; Cabellos, J.L.; Orozco-Ic, M.; Chattaraj, P.K.; Zhao, L.; Merino, G. Planar pentacoordinate carbon in CGa5+ derivatives. Phys. Chem. Chem. Phys. 2018, 20, 12350–12355.
- Zdetsis, A.D. Novel pentagonal silicon rings and nanowheels stabilized by flat pentacoordinate carbon(s). J. Chem. Phys. 2011, 134, 094312.
- Cui, Z.H.; Vassilev-Galindo, V.; Cabellos, J.L.; Osorio, E.; Orozco, M.; Pan, S.; Ding, Y.H.; Merino, G. Planar pentacoordinate carbon atoms embedded in a metallocene framework. Chem. Commun. 2017, 53, 138–141.
- Sun, R.; Jin, B.; Huo, B.; Yuan, C.; Zhai, H.J.; Wu, Y.B. Planar pentacoordinate carbon in a sulphur-surrounded boron wheel: The global minimum of CB5S5+. Chem. Commun. 2022, 58, 2552–2555.
- Erhardt, S.; Frenking, G.; Chen, Z.F.; Schleyer, P.v.R. Aromatic boron wheels with more than one carbon atom in the center: C2B8, C3B93+, and C5B11+. Angew. Chem. Int. Ed. 2005, 44, 1078–1082.
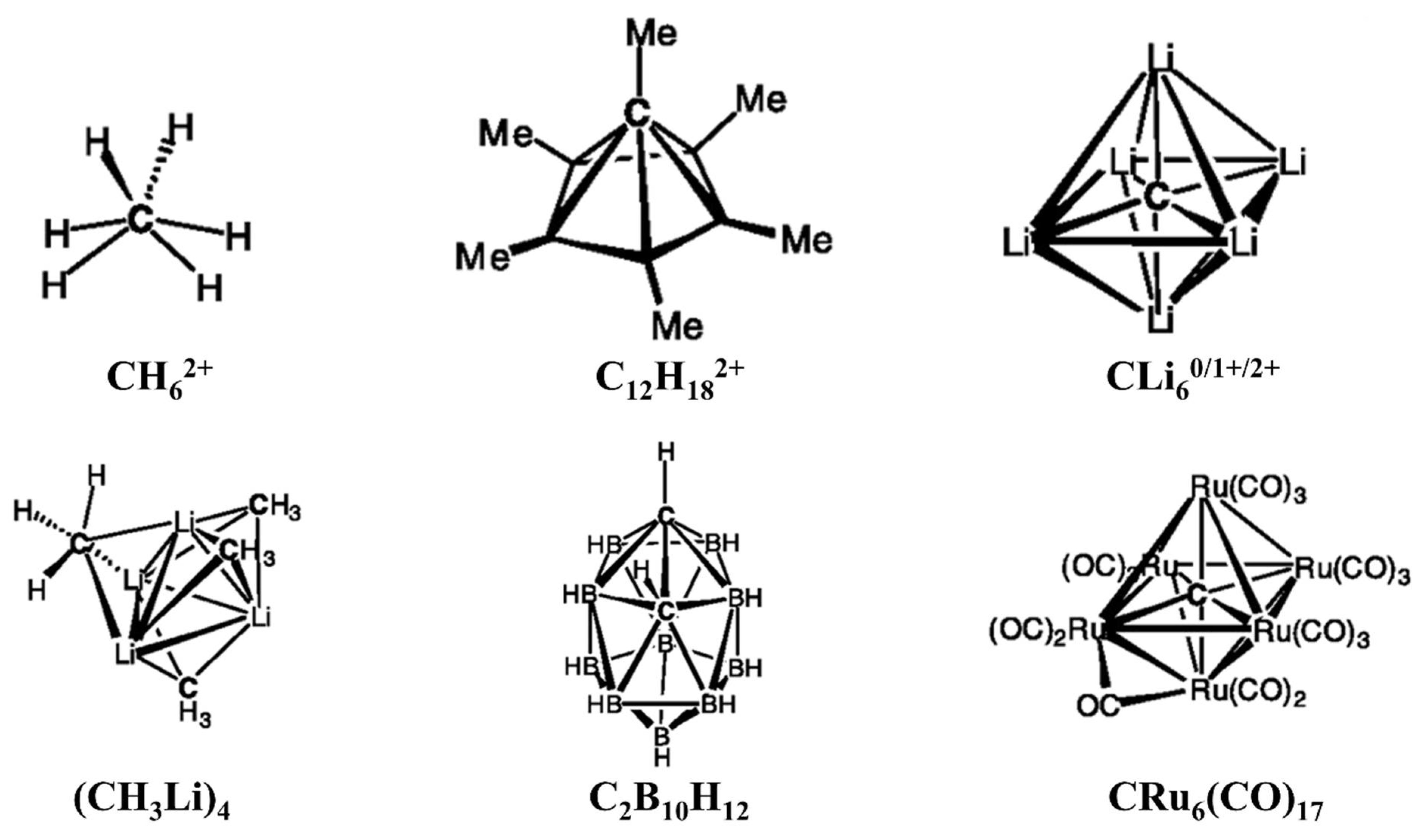
- Lammertsma, K.; Barzaghi, M.; Olah, G.A.; Pople, J.A.; Schleyer, P.v.R.; Simonetta, M. Carbodications. 7. Structure and stability of diprotonated methane, CH62+. J. Am. Chem. Soc. 1983, 105, 5258–5263.
- Sirigu, A.; Bianchi, M.; Benedetti, E. The crystal structure of Ru6C(CO)17. J. Chem. Soc. D 1969, 596a.
- Hogeveen, H.; Kwant, P.W. Pyramidal mono- and dications. Bridge between organic and organometallic chemistry. Acc. Chem. Res. 1975, 8, 413–420.
- Exner, K.; Schleyer, P.v.R. Planar Hexacoordinate Carbon: A Viable Possibility. Science 2000, 290, 1937–1940.
- Wu, Y.B.; Duan, Y.; Lu, G.; Lu, H.G.; Yang, P.; Schleyer, P.v.R.; Merino, G.; Islas, R.; Wang, Z.X. D3h CN3Be3+ and CO3Li3+: Viable planar hexacoordinate carbon prototypes. Phys. Chem. Chem. Phys. 2012, 14, 14760–14763.
- Leyva-Parra, L.; Diego, L.; Yañez, O.; Inostroza, D.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Planar hexacoordinate carbons: Half covalent, half ionic. Angew. Chem. Int. Ed. 2021, 60, 8700–8704.