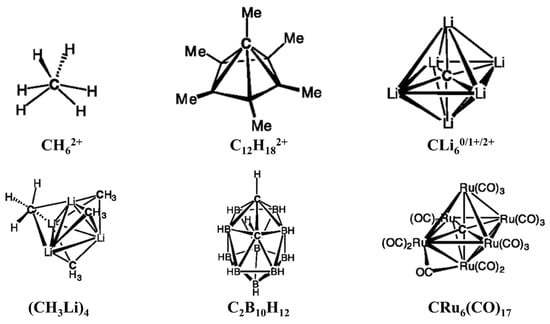
1. Planar Tetracoordinate Carbons (ptCs)
1.1. How to Achieve ptCs
Methane (CH
4) is the simplest hypothetical ptC molecule to think about. The hybridization changes from sp
3 to sp
2 for the planar
D4h configuration. The planar configuration is approximately 130 kcal mol
−1 higher in energy compared to the lowest-energy tetrahedral geometry [
12]. Even the planar structure has higher energy than the C–H bond detachment energy (103 kcal mol
−1) [
13]. After the analysis of the electronic structure of the planar CH
4 molecule, Hoffmann and co-workers suggested a way to stabilize the planar structures. When the tetrahedral structure of methane becomes planar, an extra lone pair is available on the central carbon, which distorts its planar geometry. The other point is the electron-deficient nature of the C–H bonds in the planar form. They suggested that the replacement of hydrogens by σ-donor ligands overcome the electron deficiency problem of the C–H bonds, as the ligands give electrons to the carbon atom. The ligands should have π-acceptor capacity so that they can accept the lone pair on the carbon. So, with the incorporation of simultaneous σ-donor and π-acceptor ligands, a ptC structure can be generated, and this strategy is called the “
electronic approach”. Using this approach, many ptC molecules have been theoretically reported [
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
[1],
[2],
[3],
[4]] and experimentally characterized [
32,
33,
34,
35,
36,
37,
38,
39,
40].
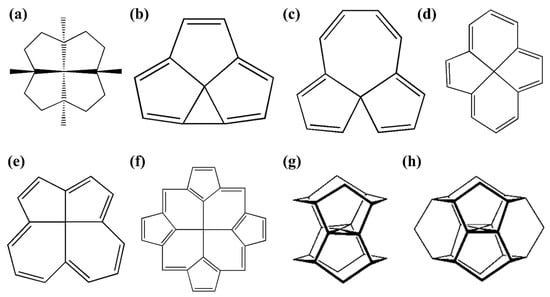
In addition to the electronic approach, ptC structures can be designed by generating enough strain to keep the carbon forcefully in planar orientations, and this method is called the “
mechanical strategy”. In this particular approach, to generate sufficient strain, cylindrical cages or tubes, small rings, and annulenes are helpful agents [
41,
42,
43,
44,
45,
46,
47]. Although some species with ptC were designed theoretically by using this strategy, no experimentally characterized ptC structures based on this strategy have been reported. Following this strategy, some of the computationally predicted ptC species are shown in
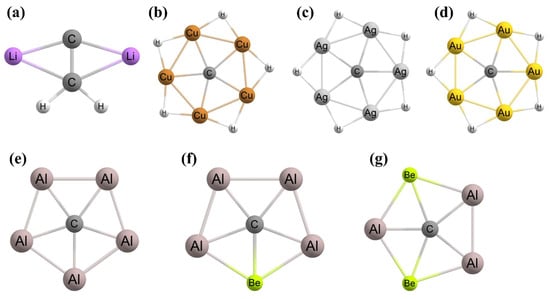
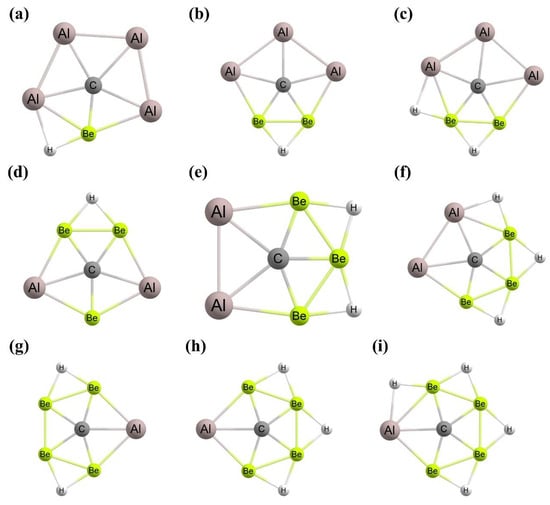
Figure 1. To achieve ptC structures based on this approach, the fenestrenes and the aromatic unsaturated fenestrenes, rigid 3D cages, such as octaplane, were proposed initially [
48,
49]. In 1999, Ding et al. noted that “
despite considerable computational efforts no structures with a planar tetracoordinate C(C)4 substructure have been found” [
50]. However, Rasmussen et al. computationally reported the first successful ptC structure based on this strategy by adjusting the
1g structure to generate
1h (
Figure 1) geometry in which the ptC atom is stabilized in a strained environment [
44,
51]. With the use of a similar strategy, Wang and Schleyer reported a set of boron spiroalkanes with a planar C(C)
4 moiety through the replacement of carbon by boron atoms [
20,
52].
Figure 1. Schematic presentations of various mechanically stabilized ptC molecules (a–h).
1.2. Early Examples of ptCs
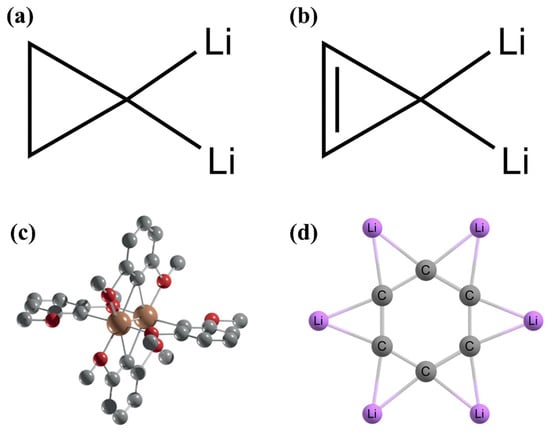
With the help of the two suggested strategies, the first example of ptC came from Collins et al. in 1976 [
53]. Through a systematic computational investigation, they reported 1,1-dilithiocyclopropane (
Figure 2a) and 3,3-dilithiocyclopropene (
Figure 2b) systems with ptCs in the energy minimum geometries and the tetrahedral orientations have higher energies than the corresponding planar configurations. One year later, the first experimentally characterized ptC containing compound V
2(2,6-dimethoxyphenyl)
4 (
Figure 2c) was published by Cotton et al. [
54]. This complex contains triple bonds between two vanadium (V) centers and has two ptCs at two ligand rings. However, unfortunately, at that time, the original authors did not realize this beautiful fact. This system has importance in this background as it is the first experimentally predicted ptC-containing compound. Due to the ionic bonding nature of lithium (Li), it prefers bridging positions, and with this concept, Xie et al. in 1991 designed a
D6h symmetric C
6Li
6 system with ptCs (
Figure 2d) [
55]. The simplest molecule with a ptC has only five atoms, and the first example of this category was CAl
2Si
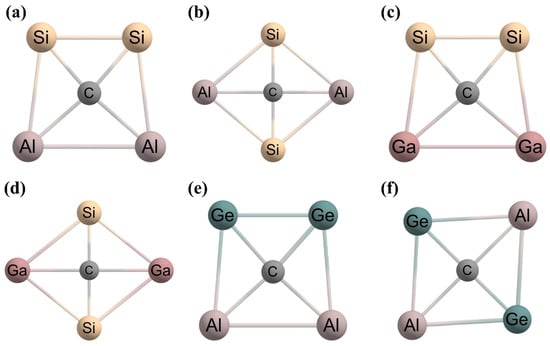
2, which was reported in 1991 by Schleyer and Boldyrev [
15]. They concluded that
cis and
trans isomers of CSi
2Al
2 (
Figure 3a and 3b, respectively) were local minimum geometries with a ptC, but they did not mention the energy of the tetrahedral-like geometry of this system. They first introduced the 18-valence electron counting concept for the stabilization of planar geometries. After seven years, in 1998, Boldyrev and Simons computed the energies of the planar and tetrahedral-like geometries of the CSi
2Al
2 system, and then they expanded the search for the possibility of a ptC atom in higher analogues such as CSi
2Ga
2 and CGe
2Al
2 species in order to determine the size dependency of the surrounding atoms on the stabilities of these species in planar orientations [
16]. The optimized structures of CSi
2Al
2 in singlet states are given in
Figure 3. The
cis and
trans isomers of the CSi
2Al
2 system are energy minima based on the theoretical analysis using the B3LYP/6-311+G* method, which is in agreement with the earlier conclusion at the MP2/6-31G* level of theory. However, both these isomers become saddle points in the MP2(full)/6-31+G* and MP2(full)/6-311+G* methods [
16]. The authors reported that in the case of the CSi
2Al
2 system, the tetrahedral-type geometry is a first-order saddle point in the B3LYP/6-311+G* and MP2(full)/6-311+G* methods and has almost 27−28 kcal mol
−1 higher energy than the more stable quasi planar
cis and
trans isomers
3a and
3b, respectively (
Figure 3) [
16]. Then they studied two 18-valence isoelectronic species, CSi
2Ga
2 and CGe
2Al
2, where the cavities for the carbon center happened to be larger than that in the CSi
2Al
2 system. However, this time they only optimized planar
cis and
trans and tetrahedral geometries for CSi
2Ga
2 and CGe
2Al
2 systems. In the B3LYP/6-311+G* and MP2(fc)/6-311+G* methods, the planar
cis and
trans isomers of both these species were reported as energy minima [
16]. However, the
cis isomer (
Figure 3c) of CSi
2Ga
2 is less stable than the
trans isomer (
Figure 3d) by 2 kcal mol
−1, while, for the CGe
2Al
2 system, the
cis isomer (
Figure 3e) has 3 kcal mol
−1 lower energy than the
trans isomer (
Figure 3f). They also reported that the tetrahedral-type geometries have 27 and 25 kcal mol
−1 more energy than the most stable isomers for CSi
2Ga
2 and CGe
2Al
2 systems, respectively. The increase in the size of the cavity in the CSi
2Ga
2 and CGe
2Al
2 species permits the incorporation of carbon into it and thus maintains the planar structures. From these analyses, they concluded that pentatomic species with a carbon center and two Al or Ga and two Si or Ge surrounding atoms should have stable planar geometries [
16]. The planar structures may be preferred over the tetrahedral one when Jahn−Teller distortion makes the tetrahedral geometries unstable and the formation of the maximal carbon–ligand and ligand−ligand bonding by the valence electrons. For this purpose, they compared the occupancy pattern of the valence molecular orbitals (MOs) of the tetrahedral CF
4 molecule with the tetrahedral structures of their systems. The CF
4 molecule is a 32-valence electronic system, and the occupancy pattern of the occupied MOs is 1a
121t
262a
122t
261e
43t
261t
16. They assumed other tetrahedral molecules or nearly tetrahedral structures will follow this occupancy pattern (except for symmetry-imposed degeneracies) and the 18-valence electronic tetrahedral structures show 1a
121t
262a
122t
261e
2 pattern of occupancy. Due to this partially filled e-orbital, the tetrahedral structures of their systems show Jahn–Teller instability and become distorted to a planar structure. They suggested that the presence of 18-valence electrons is crucial for planar geometries to be stable and preferred over tetrahedral structures. The formation of three σ and one π bonds among the middle carbon and the surrounding atoms and one ligand–ligand bond are the consequences of 18-valence electrons, the appeasement case for planar geometries. It took a long time to encourage experimental researchers to test this prognosis. Nevertheless, the species CAl
4− and CAl
3Si
− (isoelectronic to CSi
2Ga
2) were prepared in molecular beams and the planarity of these systems was experimentally confirmed [
18,
40,
56].
Figure 2. Optimized geometries of (a) 1,1-dilithiocyclopropane and (b) 3,3-dilithiocyclopropene systems. (c) The molecular structure of V2(2,6-dimethoxyphenyl)4 features a V≡V triple bond and two planar tetracoordinate carbon (ptC) centers. The H atoms are omitted for clarity. (d) Optimized D6h symmetric structure of C6Li6.
Figure 3. Optimized geometries of (a) cis-CSi2Al2, (b) trans-CSi2Al2, (c) cis-CSi2Ga2, (d) trans-CSi2Ga2, (e) cis-CGe2Al2, and (f) trans-CGe2Al2 systems. Structure (a) has 1.16 kcal mol−1 lower energy than structure (b). Structure (c) has 2.01 kcal mol−1 higher energy than structure (d). Structure (e) has 2.87 kcal mol−1 lower energy than structure (f).
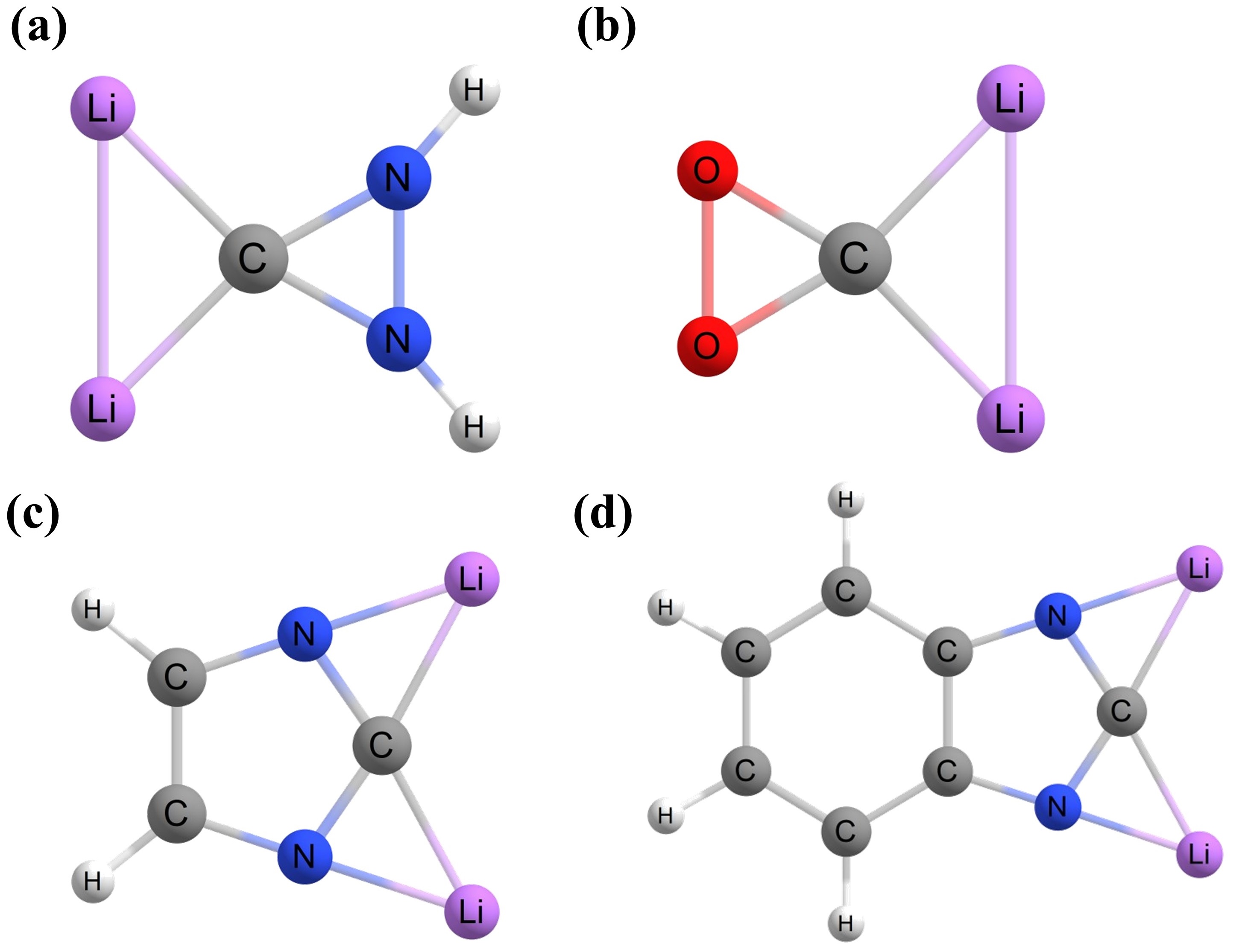
With the use of Li as ligand atoms, a series of ptC species can be adapted [
19]. For example, the replacement of the CH
2 group in the 1,1-dilithiocyclopropane (
Figure 2a) molecule by isoelectronic NH and O generates ptC structures
4a and
4b, respectively (
Figure 4). The
4c and
4d ptC structures are also generated by attaching
4a to a heterocyclic system and a benzene ring, respectively. These
4c and
4d species are more viable targets due to the dominance of the aromaticity of imidazole.
Figure 4. Optimized geometries of (a–d) computationally predicted dilithium ptC compounds.
2. Planar Pentacoordinate Carbons (ppCs)
The idea of ptC is expanded to the probability and representation of systems with ppC centers [
100]. In 1995, Bolton et al. reported the singlet 1,1-dilithioethene molecule (
Figure 5a) with the help of the ab initio quantum mechanical methods, which show the ppC local minimum structure having 7.2 kcal mol
−1 more energy compared to the lowest-energy isomer with a ptC atom [
101]. The barrier height for the interconversion between the ppC and the lowest-energy ptC isomers is approximately 0.4 kcal mol
−1 indicating that it is impossible to characterize the ppC isomer experimentally.
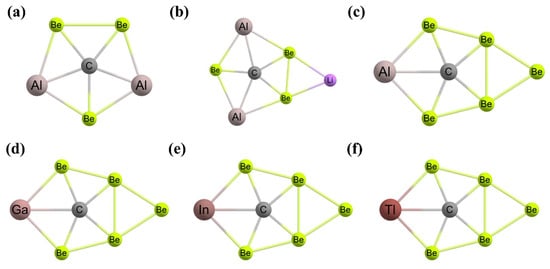
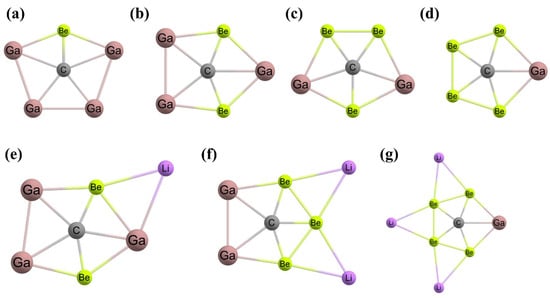
Figure 5. (a) A C2v-symmetric ppC-containing isomer of 1,1-dilithioethene contains two bridging Li centers. The computed structures of (b) Cu5H5C, (c) Ag5H5C, and (d) Au5H5C each feature bridging hydrides and a ppC. The global minimum energy structures of (e) CAl5+, (f) CAl4Be, and (g) CAl3Be2− clusters.
After the detection of the aromatic M
5(
μ-H)
5 hydrometal rings (M = Cu, Ag, Au) [
103,
104], Li et al. placed one carbon atom at the center of the Cu
5(
μ-H)
5 ring and found a perfect
D5h symmetric true local minimum structure with a ppC (
Figure 5b) [
105]. Although the Cu
5(
μ-H)
5 ring is aromatic in nature, the inclusion of the carbon causes the nonaromaticity of the ring. Further, the Ag
5H
5C (
Figure 5c) and Au
5H
5C (
Figure 5d) molecules also have a ppC in their local minimum geometries [
106].
After one year, Wang and coworkers substituted the Al atoms with isoelectronic Be atoms to generate neutral CAl
4Be (
Figure 5f) and mono-anionic CAl
3Be
2− (
Figure 5g) systems containing ppCs in the global minimum structures [
108]. The aluminum–carbon bond distances are between 2.08 and 2.29 Å, which are somewhat lengthy compared to the value of the normal aluminum–carbon bond distance of 2.00 Å but close to the 2.12 Å values as predicted theoretically for the CAl
5+ system. The bonds among the central carbon and the surrounding Be and Al atoms are longer by 0.08 Å to 0.29 Å than the corresponding normal values. The molecular dynamics simulation suggested the dynamic stability of these global minimum ppC isomers. The highly negative charges on the carbon are the consequences of the σ-donation from the surrounding atoms. At the same time, the carbon lone pair is donated to the ligand atoms. Therefore, the central carbon in the global minimum structures acts as the σ-acceptor and π-donor. The energy differences between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are 2.6 eV for both ppC structures indicating the greater stability of these isomers [
108].
Wu et al., in 2012, reported di-anionic CAl
2Be
32− (
Figure 6a) and its mono-anionic salt complex LiCAl
2Be
3− (
Figure 6b) systems by further replacement of Al atoms by Be atoms [
109]. The PES search shows the global minimum geometry of these clusters has a ppC. The vertical detachment energy (VDE) for the CAl
2Be
32− cluster is negative (−1.10 eV) indicating that this species is unstable toward electron release. However, the instability of this cluster is resolved by adding one Li
+ to neutralize one negative charge, and the most preferable binding site of Li
+ is the Be–Be bond in the LiCAl
2Be
3− cluster. In the LiCAl
2Be
3− system, the first computed VDE is 2.28 eV, indicating that the automatic removal of the additional electron is partially eliminated. Moreover, the energy gap between the HOMO and LUMO increases from 0.94 eV in CAl
2Be
32− to 1.79 eV in the LiCAl
2Be
3− system. Hence, from these stability comparisons, it is clear that LiCAl
2Be
3− system is more achievable than the CAl
2Be
32−cluster [
109]. The dynamical stability of the global minimum structures was confirmed at 4 K and 298 K temperatures up to 100 ps simulation time. The natural charge analyses indicate that the addition of Li
+ to the CAl
2Be
32− system influences the ionic bonding, but the covalent bonds are not markedly affected.
Figure 6. The global minimum energy structures of (a) CAl2Be32−, (b) LiCAl2Be3−, (c) CBe5Al−, and (d) CBe5Ga− clusters. The local minimum structures of (e) CBe5In− and (f) CBe5Tl− clusters.
Further, the complete substitution of Al atoms was performed by Castro et al., in which they generated heptaatomic anionic clusters CBe
5E
− (E = Al, Ga, In, Tl) and searched the PES of the designed systems [
110]. For CBe
5Al
− (
Figure 6c) and CBe
5Ga
− (
Figure 6d) clusters, the ppC structures are the global minima. However, for CBe
5In
− (
Figure 6e) and Cbe
5Tl
− (
Figure 6f) clusters, the ppC local minima have only 1.2 kcal mol
−1 and 1.8 kcal mol
−1 higher energy compared to the lowest-energy isomers, respectively. The geometries show that the ppC atom is present in the middle of the Be
4E ring, and an extra Be atom is bonded to one Be–Be bond in the plane. In the case of the CBe
5Al
− geometry, the aluminum–carbon bond distance is 2.233 Å, which is somewhat lengthier than the normal aluminum–carbon bond length of 2.00 Å and the 2.12 Å values theoretically predicted for the CAl
5+ system. The highly negative charges on the carbon are the consequences of the σ-donation from the surrounding atoms. From the valence electronic configuration of the carbon atom, they concluded that the carbon lone pair is donated to the ligand atoms, which assists in stabilizing the planar geometries [
110]. The authors found that the planar global minimum geometries are σ- and π-aromatic.
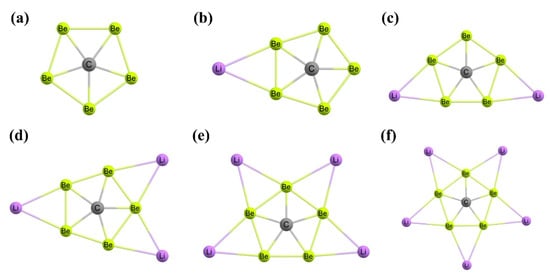
In 2008, Qiong et al. designed CBe
5 and CBe
54− systems having a ppC atom in their stable local minimum structures (
Figure 7a) [
111]. The Be
5 ring assists as a σ-donor and a π-acceptor in the
D5h structure of CBe
5 and CBe
54− systems, respectively. The NICS calculations predicted that in the
D5h structure of CBe
5 and CBe
54− systems, σ-aromaticity and π-aromaticity, respectively, are dominant. Although the CBe
54− cluster has a ppC, the greater charge density makes it unstable. However, the instability of this cluster is resolved by adding Li
+ ions to neutralize the negative charges and the resulting species are CBe
5Li
nn−4 (
n = 1 to 5) (
Figure 7b to 7f, respectively) [
112]. In these clusters, the ppC cores are preserved when Li
+ ions are bonded with their corresponding anions. The central carbon in the global minimum structures acts as the σ-acceptor and π-donor. The electron delocalization within the CBe
5Li
nn−4 (
n = 1 to 5) clusters is predicted from the induced magnetic field analysis. The energy gap between the HOMO and LUMO increases moderately upon increasing the counter ions from CBe
5Li
3− (3.47 eV) to CBe
5Li
5+ (7.11 eV), suggesting increased stability following the maximum hardness principle (MHP) [
[9],
[10]]. The molecular dynamics simulations of the systems at 1000 K for 20 ps show that the CBe
5 pentagon remains intact during the entire simulation. The negative charge density on the CBe
54− cluster is also decreased by capping H
+ ions to the system, just as in the case of Li
+ ions, and the species are CBe
5H
nn−4 (
n = 2–5) [
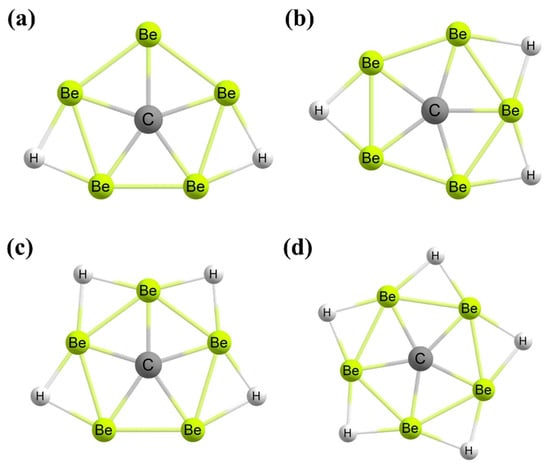
113]. In the case of CBe
5H
22− (
Figure 8a) and CBe
5H
3− (
Figure 8b) clusters, the ppC structures are the lowest-energy
C2v point group of symmetry. For the CBe
5H
4 cluster (
Figure 8c), a quasi-planar geometry has 1.8 kcal mol
−1 more energy compared to the tetrahedral global minimum structure. Moreover, the CBe
5H
5+ cluster (
Figure 8d) has a quasi-planar ppC structure as the global minimum. The stability of the ppC- or quasi-ppC-containing geometries is governed by the presence of the peripheral three-centered-two-electron Be–H–Be bonds, the origination of the stable eight-electron shell structure, and the presence of the 6σ and 2π dual aromaticity. The excess charge density on the CBe
54− system is also reduced by complexing with halogen cations (F
+, Cl
+, and Br
+) or alkali metal cations (Li
+, Na
+, and K
+) to generate CBe
5X
5+ systems [
114]. The PES search shows that the global minima of the clusters are either in ppC or quasi-ppC forms. The global minima of CBe
5F
5+, CBe
5Li
5+, CBe
5Na
5+, and CBe
5K
5+ clusters have excellently planar and extremely symmetric
D5h geometries, whereas CBe
5Cl
5+ and CBe
5Br
5+ clusters experience slight non-planar contortion as
C2 geometries. Again, in these systems, the three-centered-two-electron Be–X–Be bonds provide stability in planar forms. The NICS analysis proved the double aromatic character (σ- and π-aromaticity) of the systems is in agreement with the adaptive natural density partitioning (AdNDP) analyses [
[11],
[12]]. The molecular dynamics simulations suggested that the ppC-containing CBe
5 ring in the minimum energy structures is well conserved throughout the whole simulation, indicating that the geometries are rigid against isomerization and decomposition.
Figure 7. (a) The symmetric structures CBe5 and CBe54− are ppC species. The neutral and tetraanionic forms have the same shape but different bond lengths. The global minimum structures of (b) CBe5Li3−, (c) CBe5Li22−, (d) CBe5Li3−, (e) CBe5Li4, and (f) CBe5Li5+ clusters.
Figure 8. The global minimum structures of (a) CBe5H22− and (b) CBe5H3− clusters with a perfect ppC. The local and global minimum structures of (c) CBe5H4 and (d) CBe5H5+ clusters, respectively, with a quasi-ppC.
In 2018, Zhao et al. reported various ppC systems by adding hydrogen atoms to the CAl
4Be, CAl
3Be
2−, CAl
2Be
32−, and CAlBe
43− parent molecules [
115]. They reported nine new planar and quasi-planar ppC clusters of CAl
nBe
mH
xq (
n +
m = 5,
q = 0, ±1,
x =
q +
m − 1) (
Figure 9). The ppC core remains unchanged geometrically and electronically with the gradual introduction of hydrogen atoms. Interestingly, the energy gap between the HOMO and LUMO increases in the studied clusters as compared to the parent anionic clusters. The presence of the three-centered-two-electron Be–H–Be or Be–H–Al π bonds is responsible for the stabilization of the ppC geometries [
115]. Remarkably, among the nine studied clusters, seven molecules show ppC in the global minimum structures. Again, among the global minimum geometries, only CAl
3Be
2H, CAl
2Be
3H
−, CAl
2Be
3H
2, and CAlBe
4H
4+ clusters are dynamically stable enough. For the CAl
4BeH
4+ and CAl
3Be
2H
2+ clusters, the ppC isomers have 10.7 and 3.8 kcal mol
−1 higher energy, respectively, with respect to the lowest-energy structures. However, the closest isomers of the CAl
3Be
2H, CAl
2Be
3H
−, CAl
2Be
3H
2, CAl
2Be
3H
3+, CAlBe
4H
2−, CAlBe
4H
3, and CAlBe
4H
4+ clusters have 4.6, 3.1, 4.6, 3.6, 3.7, 2.8, and 15.0 kcal mol
−1 higher energies, respectively, with respect to the global minimum structures [
115]. The NICS calculations predicted that the considered clusters are σ- and π-dual aromatic. The natural bond orbital (NBO) computations predicted that there is a significant contribution of the ionic and covalent bonding toward the stabilization of the ppC structures.
Figure 9. Optimized structures of (a) CAl4BeH4+, (b) CAl3Be2H, (c) CAl3Be2H2+, (d) CAl2Be3H−, (e) CAl2Be3H2, (f) CAl2Be3H3+, (g) CAlBe4H2−, (h) CAlBe4H3, and (i) CAlBe4H4+ clusters.
Recently, Pan et al. reported a family of systems with ppC based on the next heaviest analogue of the CAl
5+ system [
116]. As the size of the Ga atom is larger than that of the Al, no ppC isomer is found as a global and/or local minimum for the CGa
5+ system. Hence, with the use of the smaller-sized beryllium (Be) atoms, the isoelectronic substitution of Ga atoms generated CGa
4Be, CGa
3Be
2−, CGa
2Be
32−, and CGaBe
43− clusters with ppC in the global minimum structures (
Figure 10). For the neutralization of the anionic clusters, one, two, and three Li
+ ions were used for CGa
3Be
2−, CGa
2Be
32−, and CGaBe
43− clusters, respectively, to generate CGa
3Be
2Li, CGa
2Be
3Li
2, and CGaBe
4Li
3 clusters (
Figure 10). Although the anionic systems have ppC in the global minimum structures, the first ionization potential of CGa
2Be
32− and CGaBe
43− clusters are negative (−2.91 and −6.45 eV, respectively) suggesting the spontaneous loss of an electron from the clusters [
116]. However, the first ionization potential of the CGa
3Be
2− cluster is positive (1.20 eV) indicating its stability towards the spontaneous loss of an electron. The central carbon in the global minimum structures acts as the σ-acceptor and π-donor. Moreover, the extent of π-back-bonding from the 2
pz orbital of the central carbon also increases by increasing the number of counter-ions. The electron delocalization within the system is well understood from the molecular orbitals and their magnetic responses studies. The magnetic responses indicated the σ- and π-aromaticity of the global minimum structures, and the σ-contribution is the governing one [
116].
Figure 10. The lowest-energy ppC forms for (a) CGa4Be, (b) CGa3Be2−, (c) CGa2Be32−, (d) CGaBe43−, (e) CGa3Be2Li, (f) CGa2Be3Li2, and (g) CGaBe4Li3 clusters.
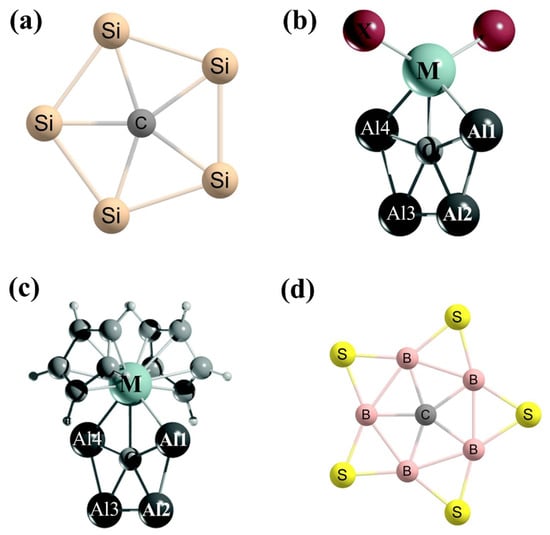
Using silicon (Si) as the surrounding atoms, Zdetsis et al. in 2011 designed Si
5C
2− and Si
5C
− clusters (
Figure 11a) with the help of the DFT and the coupled-cluster theory that predicted planar structures of the clusters stabilized by the C–Si bonds [
117]. These local minimum structures have 12.9 and 22.8 kcal mol
−1 higher energy compared to the three-dimensional-type global minimum for Si
5C
2− and Si
5C
− clusters, respectively. For the Si
5C
− cluster, the authors reported two planar structures with
D5h and
Cs point groups of symmetries that are related by Jahn–Teller distortions. However, these two geometries differ by only 8 kcal mol
−1 of energy.
Figure 11. (a) The symmetric structures Si5C2− and Si5C− clusters are ppC species. The monoanionic and dianionic forms have the same shape but different bond lengths. The lowest-energy isomers of (b) CAl4MX2 clusters and (c) CAl4M(C5H5)2 clusters. The global minimum structure of (d) CB5S5+ cluster with a ppC.
Merino and co-workers used transition metals along with the main group elements to design a ppC in the global minimum structure. They reported CAl
4MX
2 clusters (M = Zr and Hf; X = F–I and C
5H
5) with a ppC connected to a transition metal and attached to a metallocene skeleton (
Figure 11b,c) [
118]. The natural charge analysis suggested that a significant amount of electron transfer occurs from the surrounding atoms to the central carbon atom and the values are in the range of −2.21 to −2.37 |e|. The BOMD simulations assist the kinetic stability of the Zr systems at 700 K.
Very recently, Sun et al. designed a sulfur-surrounded boron wheel CB
5S
5+ cluster with a ppC in the global minimum structure (
Figure 11d) [
119]. In this cluster, the presence of the strong π back-donation from the five-bridged sulfur atoms to the boron atoms lowers the electron-deficient nature of the boron centers. The second-lowest isomer with a ptC atom has 1.1 kcal mol
−1 higher energy than the lowest-energy ppC isomer. The structure with planar pentacoordinate boron (ppB) has 61.2 kcal mol
−1 higher energy than the ppC structure. The BOMD simulation suggested that at 4 K, the global minimum is dynamically very rigid. Moreover, the planarity of the cluster is well conserved at 298 K, 500 K, and 1000 K temperatures. The considered cluster has a 7.47 eV energy gap between HOMO and LUMO, a high VDE value of 13.22 eV, and a low vertical electron affinity (VEA) of 4.31 eV, indicating an electronically robust structure [
119]. The NICS computations show the σ + π double aromaticity in the CB
5S
5+ cluster.
All the above-mentioned global and/or local minimum structures contain one ppC. In 2005, Schleyer and coworkers reported certain fluxional wheel-like species, namely, C
2B
8, C
3B
93+, and C
5B
11+, in which the interior C
2, C
3, and C
5 fragments revolve within the boron rings, respectively (
Figure 12) [
120]. In C
2B
8, C
3B
93+, and C
5B
11+ species, there are two, three, and five ppC centers that coexist in the energy minimum geometries, respectively. The NICS computations predict the π-aromaticity of these species with more than one ppC atom. These unusual planar clusters are stable when the constituent elements are suited nicely, both geometrically and electronically.
Figure 12. The boron–carbon clusters (a) C2B8, (b) C3B93+, and (c) C5B11+ are local minimum structures that include conformationally dynamic C2, C3, and C5 units, respectively, within boron rings.
3. Planar Hexacoordinate Carbons (phCs)
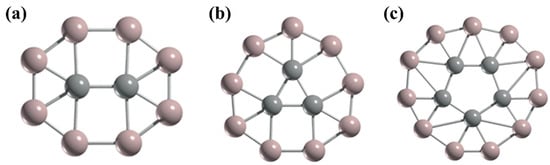
The possibility of the ptC and ppC molecules and/or ions are discussed in the previous two sections. Now the question arises of whether it is feasible to obtain a planar hexacoordinate carbon (phC) or a planar heptacoordinate carbon, or a planar octacoordinate carbon. In the case of the main group of components, the existence of planar hexacoordination is limited. Despite various species with hexacoordinate carbon being described, they have 3D geometries (
Figure 13) [
121,
122,
123]. In 2000, the first example with a phC is the CB
62− di-anionic system (
Figure 14a), studied by Exner et al. using DFT and high-level ab initio calculations [
124]. This system has a
D6h point group of symmetry. The reported structure is not the lowest-energy isomer, rather it is a local minimum with 143.9 kJ mol
−1 more energy compared to the lowest-energy structure. This cluster shows benzene-like HOMOs and is aromatic in nature.
Figure 13. Some compounds with hexacoordinate carbons have three-dimensional structures.
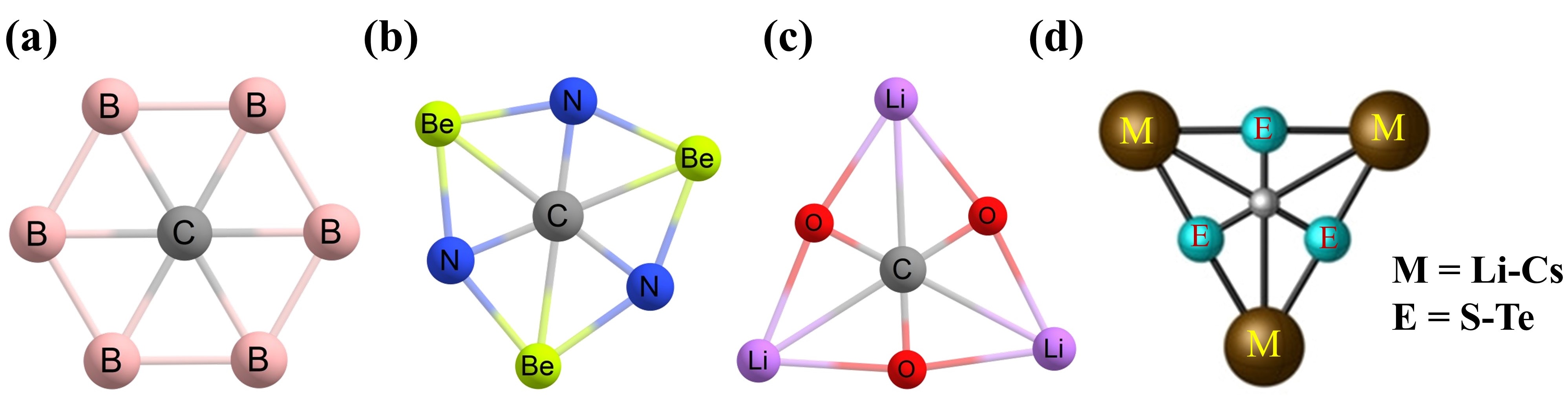
Figure 14. Local minimum structures of (a) CB62− and (b) CN3Be3+ clusters with a phC. Global minimum structures of (c) CO3Li3+ and (d) CE3M3+ (E = S–Te, and M = Li–Cs) clusters.
In 2012, Wu et al. used a CB
62− unit and executed isoelectronic replacement on it to compose unipositive CN
3Be
3+ and CO
3Li
3+ clusters (
Figure 14b and 14c, respectively) [
126]. Both clusters in their phC structures correspond to the
D3h symmetry. The CN
3Be
3+ cluster is a local minimum with 25.5 kJ mol
−1 more energy with respect to the global minimum geometry. However, the CO
3Li
3+ cluster is a putative global minimum with a phC center. The authors mentioned that the three bridging Li
+ ions stabilized the CO
32− ion electrostatically. Later, Leyva-Parra et al. studied the charges on the middle carbon and the bridging Li centers, and the values are 0.87 |e| and 0.97 |e|, respectively [
127]. Hence, the positive charges on both carbon and lithium atoms imply electrostatic repulsion between them. Due to this repulsion and the nonappearance of any remarkable orbital overlap between them, this hexacoordinate environment is ambiguous. Because of the different electronegativity values of carbon and oxygen atoms, positive charges on the phC are expected. Therefore, Leyva-Parra et al. substituted the oxygen atoms with the least electronegative sulphur atoms to obtain a true phC atom in the global minimum structures [
127]. The fifteen possible CE
3M
3+ (E=S–Te, and M=Li–Cs) combinations are to be identified as phC clusters (
Figure 14d). The natural charge analysis shows negative charges on the phC and positive charges on the bridging metal atoms suggesting electrostatic attractions between the partially negative central carbon atom and partially positive bridged metal atoms. These phC clusters were designed following the so-called “
proper polarization of ligand” approach [
127]. Through systematic bonding analyses, the authors stated the covalent nature of the C–E bonds and the ionic nature of the C–M bonds.
This entry is adapted from the peer-reviewed paper 10.3390/chemistry4040113