T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive subtype of hematological malignancy characterized by its high heterogeneity and potentially life-threatening clinical features. It has shown the indispensable effects of leukemia-initiating cells (LICs) and leukemic niches on T-ALL initiation and progression. These milestones greatly facilitate precision medicine by interfering with the pathways that are associated with LICs and leukemic niches or by targeting themselves directly. Most of these novel agents, either alone or in combination with conventional chemotherapy, have shown promising preclinical results, facilitating them to be further evaluated under clinical trials.
- T-cell acute lymphoblastic leukemia
- leukemia-initiating cell
- microenvironment
- leukemic niche
1. Introduction
2. LICs in T-ALL
2.1. T-LICs in Mouse Models
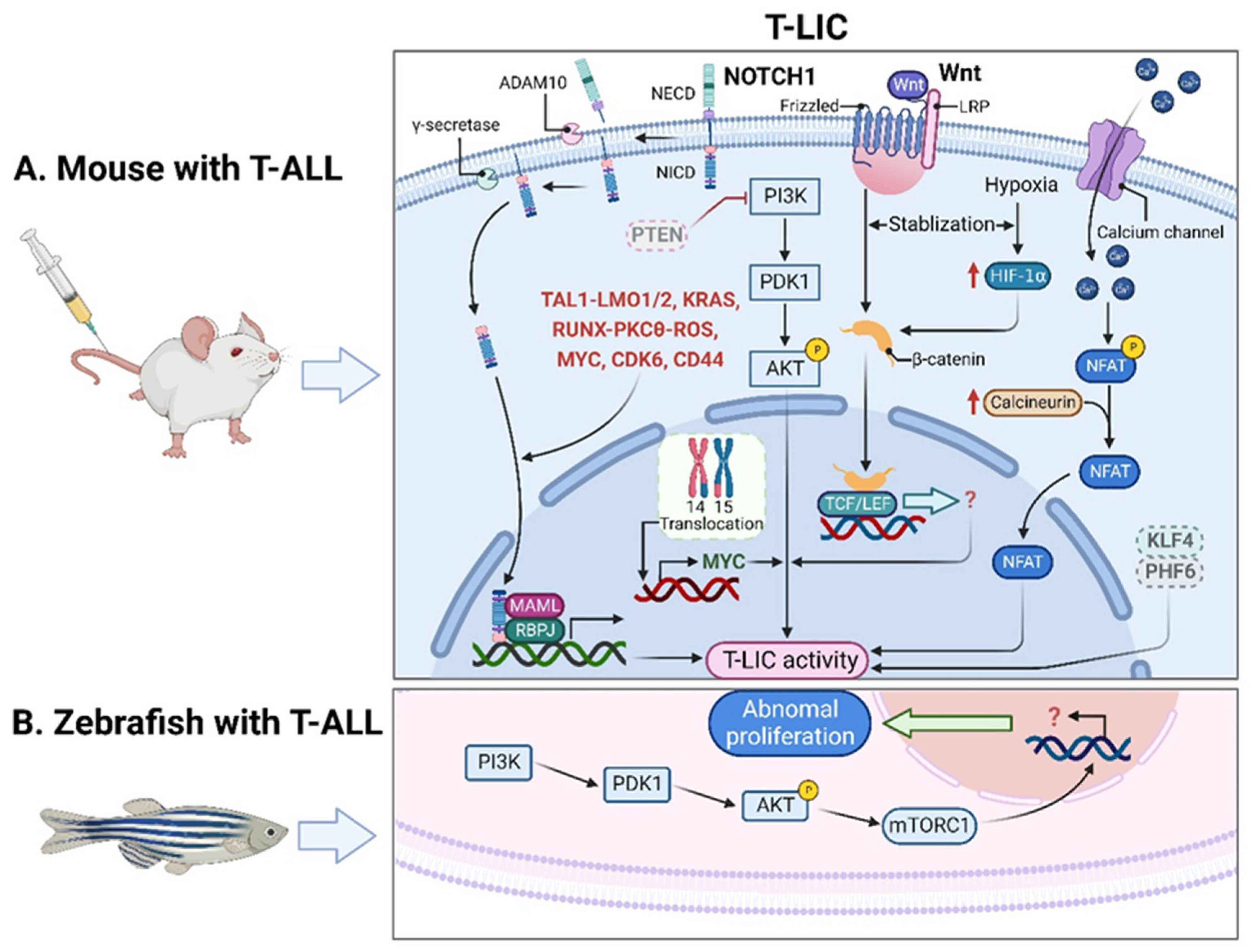
2.2. T-LICs in Zebrafish Models
2.3. Therapies Targeting T-LICs
3. Leukemic Niches in T-ALL
3.1. Effects of Leukemic Niches on T-ALL Cells
3.1.1. CXC Chemokine Ligand 12 (CXCL12)/CXC Chemokine Receptor 4 (CXCR4) Signaling
CXCL12 (also termed stromal cell derived factor-1, SDF-1) is a chemotactic factor, and its receptor CXCR4 is a highly conserved G-protein-coupled seven transmembrane receptor. Since stromal-vascular BM niches secrete abundant CXCL12, whereas CXCR4 is primarily expressed on HSCs and malignant cells, the CXCL12/CXCR4 axis becomes a key pathway for both normal hematopoiesis and cancer cell homing to the BM [48][49][50][51]. Indeed, T-ALL cells express high surface levels of CXCR4 in a calcineurin-dependent manner, and CXCR4 silencing impairs migration and survival of leukemic cells, as well as T-LIC activity [52]. Likewise, CXCL12 deletion from vascular endothelial niches impedes T-ALL development [53]. Mechanistically, the interaction between CXCL12 and CXCR4 results in activation of many survival pathways including PI3K-AKT and mitogen-activated protein kinase (MAPK) signaling cascades in T-ALL cells [54] (Figure 2A). The CXCL12/CXCR4 signaling also mediates enhanced extramedullary infiltration and dissemination [55][56]. Therefore, the activation of CXCL12/CXCR4 signaling creates a sanctuary microenvironment for T-ALL cells and confers resistance to conventional therapies [54][57]. As such, inhibition of either the CXCL12/CXCR4 interaction or its downstream signaling is of therapeutic benefit in patients with chemoresistant T-ALL [58].
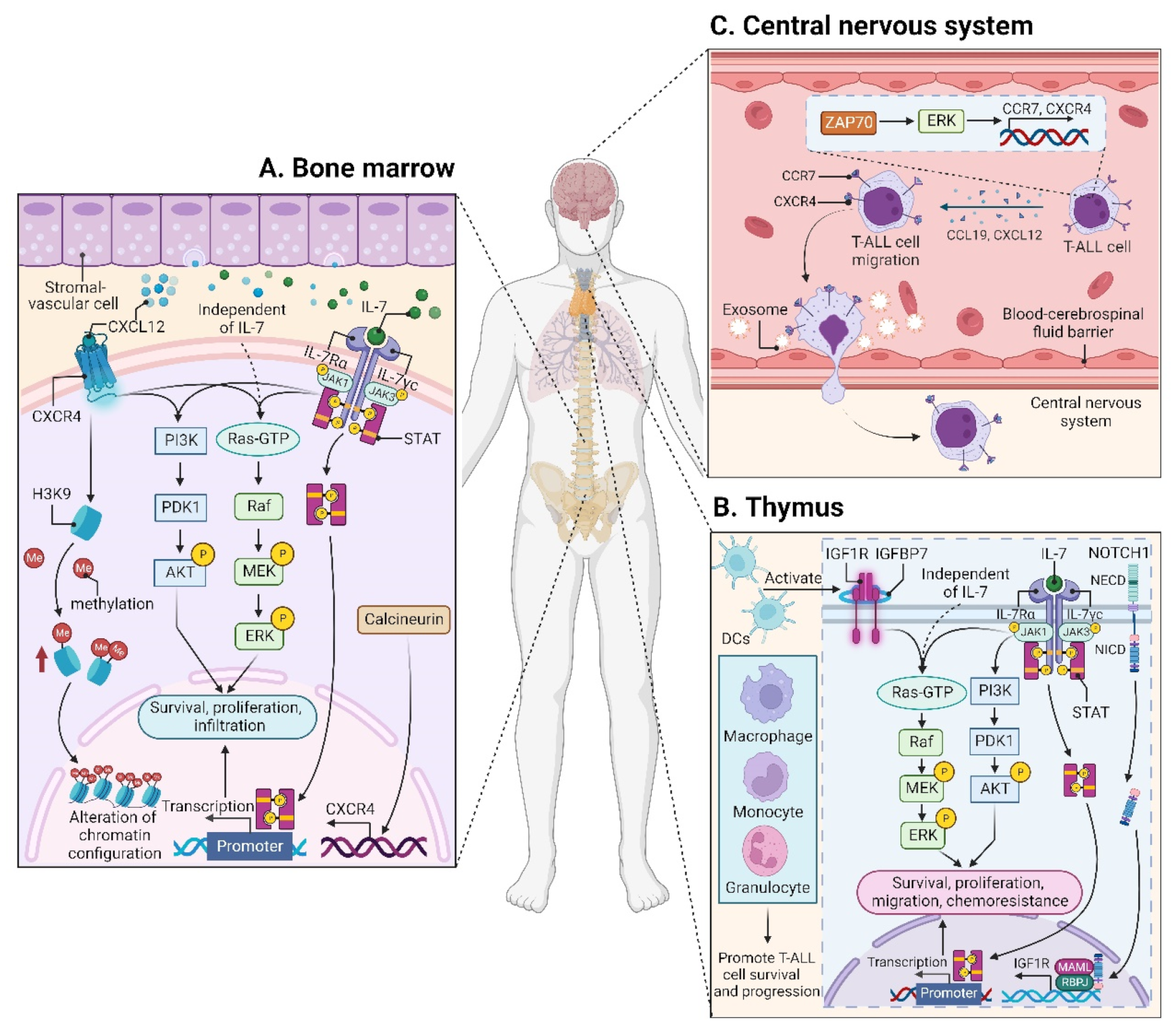
3.1.2. Insulin-like Growth Factor 1 (IGF1)/IGF1 Receptor (IGF1R) Signaling
3.1.3. Interleukin (IL7)/IL7 Receptor (IL7R) Signaling
3.1.4. CC Chemokine Ligand 19 (CCL19)/CC Chemokine Receptor 7 (CCR7) Signaling
3.2. Microenvironmental Alterations
3.2.1. BM Microenvironment
Modification of the BM microenvironment by leukemia cells was well-characterized in B-ALL and AML [83][84][85], whereas the research on the alterations of the BM microenvironment by T-ALL cells is very limited. In 2016, an elegant work reported that an accumulated T-ALL burden within the BM leads to rapid and selective remodeling of the endosteal space, resulting in a complete loss of mature osteoblasts and impairment of normal HSCs [42].
3.2.2. Thymic Microenvironment
3.2.3. Splenic Microenvironment
4. Preclinically- and Clinically-Evaluated Precision Medicine for T-ALL
4.1. Agents Targeting Aberrant Pathways
4.1.1. NOTCH1 Signaling
4.1.2. BCL2 Signaling
4.1.3. JAK-STAT Signaling
4.1.4. PI3K-AKT-mTOR Signaling
4.1.5. CDK4/6-Mediated Signaling
4.1.6. Other Signaling Pathways
4.2. Antibody-Based Therapy
4.2.1. CD38 mAbs
4.2.2. CD52 mAbs
4.2.3. IL7Rα mAbs
This entry is adapted from the peer-reviewed paper 10.3390/cancers14225655
References
- Grunenberg, A.; Sala, E.; Kapp-Schwoerer, S.; Viardot, A. Pharmacotherapeutic management of T-cell acute lymphoblastic leukemia in adults: An update of the literature. Expert Opin. Pharm. 2022, 23, 561–571.
- Fattizzo, B.; Rosa, J.; Giannotta, J.A.; Baldini, L.; Fracchiolla, N.S. The Physiopathology of T- Cell Acute Lymphoblastic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 10, 273.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648.
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737.
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291.
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322.
- Kelliher, M.A.; Seldin, D.C.; Leder, P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J. 1996, 15, 5160–5166.
- Aplan, P.D.; Jones, C.A.; Chervinsky, D.S.; Zhao, X.; Ellsworth, M.; Wu, C.; McGuire, E.A.; Gross, K.W. An scl gene product lacking the transactivation domain induces bony abnormalities and cooperates with LMO1 to generate T-cell malignancies in transgenic mice. EMBO J. 1997, 16, 2408–2419.
- Larson, R.C.; Fisch, P.; Larson, T.A.; Lavenir, I.; Langford, T.; King, G.; Rabbitts, T.H. T cell tumours of disparate phenotype in mice transgenic for Rbtn-2. Oncogene 1994, 9, 3675–3681.
- Tremblay, M.; Tremblay, C.S.; Herblot, S.; Aplan, P.D.; Hebert, J.; Perreault, C.; Hoang, T. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev. 2010, 24, 1093–1105.
- Tatarek, J.; Cullion, K.; Ashworth, T.; Gerstein, R.; Aster, J.C.; Kelliher, M.A. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood 2011, 118, 1579–1590.
- Chiang, M.Y.; Xu, L.; Shestova, O.; Histen, G.; L’Heureux, S.; Romany, C.; Childs, M.E.; Gimotty, P.A.; Aster, J.C.; Pear, W.S. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J. Clin. Investig. 2008, 118, 3181–3194.
- Giambra, V.; Jenkins, C.R.; Wang, H.; Lam, S.H.; Shevchuk, O.O.; Nemirovsky, O.; Wai, C.; Gusscott, S.; Chiang, M.Y.; Aster, J.C.; et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-theta and reactive oxygen species. Nat. Med. 2012, 18, 1693–1698.
- Chiang, M.Y.; Shestova, O.; Xu, L.; Aster, J.C.; Pear, W.S. Divergent effects of supraphysiologic Notch signals on leukemia stem cells and hematopoietic stem cells. Blood 2013, 121, 905–917.
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566.
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; Pathak, A.; Brown, N.; Deshpande, A.; et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1033–1043.
- Kong, G.; Du, J.; Liu, Y.; Meline, B.; Chang, Y.I.; Ranheim, E.A.; Wang, J.; Zhang, J. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J. Biol. Chem. 2013, 288, 18219–18227.
- Guo, W.; Lasky, J.L.; Chang, C.J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M.; et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008, 453, 529–533.
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059.
- Aifantis, I.; Raetz, E.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390.
- Grabher, C.; von Boehmer, H.; Look, A.T. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2006, 6, 347–359.
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271.
- Armstrong, F.; Brunet de la Grange, P.; Gerby, B.; Rouyez, M.C.; Calvo, J.; Fontenay, M.; Boissel, N.; Dombret, H.; Baruchel, A.; Landman-Parker, J.; et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 2009, 113, 1730–1740.
- Gerby, B.; Tremblay, C.S.; Tremblay, M.; Rojas-Sutterlin, S.; Herblot, S.; Hebert, J.; Sauvageau, G.; Lemieux, S.; Lecuyer, E.; Veiga, D.F.; et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PLoS Genet. 2014, 10, e1004768.
- Garcia-Peydro, M.; Fuentes, P.; Mosquera, M.; Garcia-Leon, M.J.; Alcain, J.; Rodriguez, A.; Garcia de Miguel, P.; Menendez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818.
- Frazer, J.K.; Meeker, N.D.; Rudner, L.; Bradley, D.F.; Smith, A.C.; Demarest, B.; Joshi, D.; Locke, E.E.; Hutchinson, S.A.; Tripp, S.; et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia 2009, 23, 1825–1835.
- Smith, A.C.; Raimondi, A.R.; Salthouse, C.D.; Ignatius, M.S.; Blackburn, J.S.; Mizgirev, I.V.; Storer, N.Y.; de Jong, J.L.; Chen, A.T.; Zhou, Y.; et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood 2010, 115, 3296–3303.
- Blackburn, J.S.; Liu, S.; Wilder, J.L.; Dobrinski, K.P.; Lobbardi, R.; Moore, F.E.; Martinez, S.A.; Chen, E.Y.; Lee, C.; Langenau, D.M. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 2014, 25, 366–378.
- Simioni, C.; Neri, L.M.; Tabellini, G.; Ricci, F.; Bressanin, D.; Chiarini, F.; Evangelisti, C.; Cani, A.; Tazzari, P.L.; Melchionda, F.; et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 2012, 26, 2336–2342.
- Roderick, J.E.; Tesell, J.; Shultz, L.D.; Brehm, M.A.; Greiner, D.L.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Gutierrez, A.; Look, A.T.; et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 2014, 123, 1040–1050.
- Medyouf, H.; Alcalde, H.; Berthier, C.; Guillemin, M.C.; dos Santos, N.R.; Janin, A.; Decaudin, D.; de The, H.; Ghysdael, J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 2007, 13, 736–741.
- Piya, S.; Yang, Y.; Bhattacharya, S.; Sharma, P.; Ma, H.; Mu, H.; He, H.; Ruvolo, V.; Baran, N.; Davis, R.E.; et al. Targeting the NOTCH1-MYC-CD44 axis in leukemia-initiating cells in T-ALL. Leukemia 2022, 36, 1261–1273.
- Grimaldi, C.; Chiarini, F.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Battistelli, M.; Falcieri, E.; Bortul, R.; Melchionda, F.; Iacobucci, I.; et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: Therapeutic implications. Leukemia 2012, 26, 91–100.
- Diamanti, P.; Cox, C.V.; Moppett, J.P.; Blair, A. Parthenolide eliminates leukemia-initiating cell populations and improves survival in xenografts of childhood acute lymphoblastic leukemia. Blood 2013, 121, 1384–1393.
- Passaro, D.; Quang, C.T.; Ghysdael, J. Microenvironmental cues for T-cell acute lymphoblastic leukemia development. Immunol. Rev. 2016, 271, 156–172.
- Passaro, D. Myeloid cells hold the master key for T-ALL spread. Blood 2020, 136, 1799–1800.
- Calvo, J.; Fahy, L.; Uzan, B.; Pflumio, F. Desperately seeking a home marrow niche for T-cell acute lymphoblastic leukaemia. Adv. Biol. Regul. 2019, 74, 100640.
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298.
- Chen, S.Y.; Yang, X.; Feng, W.L.; Liao, J.F.; Wang, L.N.; Feng, L.; Lin, Y.M.; Ren, Q.; Zheng, G.G. Organ-specific microenvironment modifies diverse functional and phenotypic characteristics of leukemia-associated macrophages in mouse T cell acute lymphoblastic leukemia. J. Immunol. 2015, 194, 2919–2929.
- Ma, S.; Shi, Y.; Pang, Y.; Dong, F.; Cheng, H.; Hao, S.; Xu, J.; Zhu, X.; Yuan, W.; Cheng, T.; et al. Notch1-induced T cell leukemia can be potentiated by microenvironmental cues in the spleen. J. Hematol. Oncol. 2014, 7, 71.
- Xiong, H.; Mancini, M.; Gobert, M.; Shen, S.; Furtado, G.C.; Lira, S.A.; Parkhurst, C.N.; Garambois, V.; Brengues, M.; Tadokoro, C.E.; et al. Spleen plays a major role in DLL4-driven acute T-cell lymphoblastic leukemia. Theranostics 2021, 11, 1594–1608.
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522.
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant Notch Signaling in the Bone Marrow Microenvironment of Acute Lymphoid Leukemia Suppresses Osteoblast-Mediated Support of Hematopoietic Niche Function. Cancer Res. 2016, 76, 1641–1652.
- Georgievski, A.; Michel, A.; Thomas, C.; Mlamla, Z.; Pais de Barros, J.P.; Lemaire-Ewing, S.; Garrido, C.; Quere, R. Acute lymphoblastic leukemia-derived extracellular vesicles affect quiescence of hematopoietic stem and progenitor cells. Cell Death Dis. 2022, 13, 337.
- Ghezzo, M.N.; Fernandes, M.T.; Pacheco-Leyva, I.; Rodrigues, P.M.; Machado, R.S.; Araujo, M.A.S.; Kalathur, R.K.; Futschik, M.E.; Alves, N.L.; Dos Santos, N.R. FoxN1-dependent thymic epithelial cells promote T-cell leukemia development. Carcinogenesis 2018, 39, 1463–1476.
- Fernandes, M.T.; Ghezzo, M.N.; Silveira, A.B.; Kalathur, R.K.; Povoa, V.; Ribeiro, A.R.; Brandalise, S.R.; Dejardin, E.; Alves, N.L.; Ghysdael, J.; et al. Lymphotoxin-beta receptor in microenvironmental cells promotes the development of T-cell acute lymphoblastic leukaemia with cortical/mature immunophenotype. Br. J. Haematol. 2015, 171, 736–751.
- Di Grande, A.; Peirs, S.; Donovan, P.D.; Van Trimpont, M.; Morscio, J.; Lintermans, B.; Reunes, L.; Vandamme, N.; Goossens, S.; Nguyen, H.A.; et al. The spleen as a sanctuary site for residual leukemic cells following ABT-199 monotherapy in ETP-ALL. Blood Adv. 2021, 5, 1963–1976.
- de Bock, C.E.; Cools, J. T-ALL: Home Is where the CXCL12 Is. Cancer Cell 2015, 27, 745–746.
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590.
- Su, L.; Hu, Z.; Yang, Y.G. Role of CXCR4 in the progression and therapy of acute leukaemia. Cell Prolif. 2021, 54, e13076.
- Mehrpouri, M. The contributory roles of the CXCL12/CXCR4/CXCR7 axis in normal and malignant hematopoiesis: A possible therapeutic target in hematologic malignancies. Eur. J. Pharmacol. 2022, 920, 174831.
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Da Costa De Jesus, C.; Lasgi, C.; Tran Quang, C.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779.
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768.
- Liou, A.; Delgado-Martin, C.; Teachey, D.T.; Hermiston, M.L. The CXCR4/CXCL12 Axis Mediates Chemotaxis, Survival, and Chemoresistance in T-Cell Acute Lymphoblastic Leukemia. Blood 2014, 124, 3629.
- Crazzolara, R.; Kreczy, A.; Mann, G.; Heitger, A.; Eibl, G.; Fink, F.M.; Möhle, R.; Meister, B. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2001, 115, 545–553.
- Jost, T.R.; Borga, C.; Radaelli, E.; Romagnani, A.; Perruzza, L.; Omodho, L.; Cazzaniga, G.; Biondi, A.; Indraccolo, S.; Thelen, M.; et al. Role of CXCR4-mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2016, 99, 1077–1087.
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine 2018, 109, 11–16.
- Lonetti, A.; Cappellini, A.; Bertaina, A.; Locatelli, F.; Pession, A.; Buontempo, F.; Evangelisti, C.; Evangelisti, C.; Orsini, E.; Zambonin, L.; et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J. Hematol. Oncol. 2016, 9, 114.
- Triplett, T.A.; Cardenas, K.T.; Lancaster, J.N.; Hu, Z.; Selden, H.J.; Jasso, G.J.; Balasubramanyam, S.; Chan, K.; Li, L.; Chen, X.; et al. Endogenous dendritic cells from the tumor microenvironment support T-ALL growth via IGF1R activation. Proc. Natl. Acad. Sci. USA 2016, 113, E1016–E1025.
- Lyu, A.; Triplett, T.A.; Nam, S.H.; Hu, Z.; Arasappan, D.; Godfrey, W.H.; Ames, R.Y.; Sarang, A.; Selden, H.J.; Lee, C.H.; et al. Tumor-associated myeloid cells provide critical support for T-ALL. Blood 2020, 136, 1837–1850.
- Scupoli, M.T.; Vinante, F.; Krampera, M.; Vincenzi, C.; Nadali, G.; Zampieri, F.; Ritter, M.A.; Eren, E.; Santini, F.; Pizzolo, G. Thymic epithelial cells promote survival of human T-cell acute lymphoblastic leukemia blasts: The role of interleukin-7. Haematologica 2003, 88, 1229–1237.
- Silva, A.; Laranjeira, A.B.; Martins, L.R.; Cardoso, B.A.; Demengeot, J.; Yunes, J.A.; Seddon, B.; Barata, J.T. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011, 71, 4780–4789.
- Ribeiro, D.; Melao, A.; van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213.
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939.
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908.
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163.
- Treanor, L.M.; Zhou, S.; Janke, L.; Churchman, M.L.; Ma, Z.; Lu, T.; Chen, S.C.; Mullighan, C.G.; Sorrentino, B.P. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J. Exp. Med. 2014, 211, 701–713.
- Canté-Barrett, K.; Spijkers-Hagelstein, J.A.; Buijs-Gladdines, J.G.; Uitdehaag, J.C.; Smits, W.K.; van der Zwet, J.; Buijsman, R.C.; Zaman, G.J.; Pieters, R.; Meijerink, J.P. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1832–1843.
- Kim, R.; Boissel, N.; Touzart, A.; Leguay, T.; Thonier, F.; Thomas, X.; Raffoux, E.; Huguet, F.; Villarese, P.; Fourrage, C.; et al. Adult T-cell acute lymphoblastic leukemias with IL7R pathway mutations are slow-responders who do not benefit from allogeneic stem-cell transplantation. Leukemia 2020, 34, 1730–1740.
- van der Zwet, J.C.G.; Buijs-Gladdines, J.; Cordo, V.; Debets, D.O.; Smits, W.K.; Chen, Z.; Dylus, J.; Zaman, G.J.R.; Altelaar, M.; Oshima, K.; et al. MAPK-ERK is a central pathway in T-cell acute lymphoblastic leukemia that drives steroid resistance. Leukemia 2021, 35, 3394–3405.
- Li, Y.; Buijs-Gladdines, J.G.; Cante-Barrett, K.; Stubbs, A.P.; Vroegindeweij, E.M.; Smits, W.K.; van Marion, R.; Dinjens, W.N.; Horstmann, M.; Kuiper, R.P.; et al. IL-7 Receptor Mutations and Steroid Resistance in Pediatric T cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study. PLoS Med. 2016, 13, e1002200.
- Silva, A.; Almeida, A.R.M.; Cachucho, A.; Neto, J.L.; Demeyer, S.; de Matos, M.; Hogan, T.; Li, Y.; Meijerink, J.; Cools, J.; et al. Overexpression of wild-type IL-7Rα promotes T-cell acute lymphoblastic leukemia/lymphoma. Blood 2021, 138, 1040–1052.
- González-García, S.; Mosquera, M.; Fuentes, P.; Palumbo, T.; Escudero, A.; Pérez-Martínez, A.; Ramírez, M.; Corcoran, A.E.; Toribio, M.L. IL-7R is essential for leukemia-initiating cell activity of T-cell acute lymphoblastic leukemia. Blood 2019, 134, 2171–2182.
- Tremblay, C.S.; Brown, F.C.; Collett, M.; Saw, J.; Chiu, S.K.; Sonderegger, S.E.; Lucas, S.E.; Alserihi, R.; Chau, N.; Toribio, M.L.; et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia 2016, 30, 1993–2001.
- Degryse, S.; de Bock, C.E.; Cox, L.; Demeyer, S.; Gielen, O.; Mentens, N.; Jacobs, K.; Geerdens, E.; Gianfelici, V.; Hulselmans, G.; et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014, 124, 3092–3100.
- de Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Vanden Bempt, M.; Dagklis, A.; et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov. 2018, 8, 616–631.
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401.
- de Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat. Commun. 2019, 10, 2517.
- Buonamici, S.; Trimarchi, T.; Ruocco, M.G.; Reavie, L.; Cathelin, S.; Mar, B.G.; Klinakis, A.; Lukyanov, Y.; Tseng, J.C.; Sen, F.; et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 2009, 459, 1000–1004.
- Alsadeq, A.; Fedders, H.; Vokuhl, C.; Belau, N.M.; Zimmermann, M.; Wirbelauer, T.; Spielberg, S.; Vossen-Gajcy, M.; Cario, G.; Schrappe, M.; et al. The role of ZAP70 kinase in acute lymphoblastic leukemia infiltration into the central nervous system. Haematologica 2017, 102, 346–355.
- Cuesta-Mateos, C.; Fuentes, P.; Schrader, A.; Juarez-Sanchez, R.; Loscertales, J.; Mateu-Albero, T.; Vega-Piris, L.; Espartero-Santos, M.; Marcos-Jimenez, A.; Sanchez-Lopez, B.A.; et al. CCR7 as a novel therapeutic target in t-cell PROLYMPHOCYTIC leukemia. Biomark. Res. 2020, 8, 54.
- Erb, U.; Hikel, J.; Meyer, S.; Ishikawa, H.; Worst, T.S.; Nitschke, K.; Nuhn, P.; Porubsky, S.; Weiss, C.; Schroten, H.; et al. The Impact of Small Extracellular Vesicles on Lymphoblast Trafficking across the Blood-Cerebrospinal Fluid Barrier In Vitro. Int. J. Mol. Sci. 2020, 21, 5491.
- Krause, D.S.; Scadden, D.T. A hostel for the hostile: The bone marrow niche in hematologic neoplasms. Haematologica 2015, 100, 1376–1387.
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, e90036.
- Kumar, R.; Godavarthy, P.S.; Krause, D.S. The bone marrow microenvironment in health and disease at a glance. J. Cell Sci. 2018, 131, jcs201707.
- Peaudecerf, L.; Lemos, S.; Galgano, A.; Krenn, G.; Vasseur, F.; Di Santo, J.P.; Ezine, S.; Rocha, B. Thymocytes may persist and differentiate without any input from bone marrow progenitors. J. Exp. Med. 2012, 209, 1401–1408.
- Martins, V.C.; Ruggiero, E.; Schlenner, S.M.; Madan, V.; Schmidt, M.; Fink, P.J.; von Kalle, C.; Rodewald, H.R. Thymus-autonomous T cell development in the absence of progenitor import. J. Exp. Med. 2012, 209, 1409–1417.
- Martins, V.C.; Busch, K.; Juraeva, D.; Blum, C.; Ludwig, C.; Rasche, V.; Lasitschka, F.; Mastitsky, S.E.; Brors, B.; Hielscher, T.; et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature 2014, 509, 465–470.
- Ballesteros-Arias, L.; Silva, J.G.; Paiva, R.A.; Carbonetto, B.; Faisca, P.; Martins, V.C. T Cell Acute Lymphoblastic Leukemia as a Consequence of Thymus Autonomy. J. Immunol. 2019, 202, 1137–1144.
- Simone, J.V.; Verzosa, M.S.; Rudy, J.A. Initial features and prognosis in 363 children with acute lymphocytic leukemia. Cancer 1975, 36, 2099–2108.
- Shuster, J.J.; Falletta, J.M.; Pullen, D.J.; Crist, W.M.; Humphrey, G.B.; Dowell, B.L.; Wharam, M.D.; Borowitz, M. Prognostic factors in childhood T-cell acute lymphoblastic leukemia: A Pediatric Oncology Group study. Blood 1990, 75, 166–173.
- Majumdar, G.; Singh, A.K. Role of splenectomy in chronic lymphocytic leukaemia with massive splenomegaly and cytopenia. Leuk. Lymphoma 1992, 7, 131–134.
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2012, 11, 1565–1575.
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350.
- Borthakur, G.; Martinelli, G.; Raffoux, E.; Chevallier, P.; Chromik, J.; Lithio, A.; Smith, C.L.; Yuen, E.; Oakley, G.J., 3rd; Benhadji, K.A.; et al. Phase 1 study to evaluate Crenigacestat (LY3039478) in combination with dexamethasone in patients with T-cell acute lymphoblastic leukemia and lymphoma. Cancer 2021, 127, 372–380.
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210.
- Lopez-Nieva, P.; Gonzalez-Sanchez, L.; Cobos-Fernandez, M.A.; Cordoba, R.; Santos, J.; Fernandez-Piqueras, J. More Insights on the Use of gamma-Secretase Inhibitors in Cancer Treatment. Oncologist 2021, 26, e298–e305.
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol. Cancer Ther. 2010, 9, 1618–1628.
- Baratta, M.G. Adjusting the focus on gamma-secretase inhibition. Nat. Rev. Cancer 2019, 19, 419.
- Habets, R.A.; de Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J.; Liston, A.; et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 2019, 11, eaau6246.
- Peirs, S.; Matthijssens, F.; Goossens, S.; Van de Walle, I.; Ruggero, K.; de Bock, C.E.; Degryse, S.; Cante-Barrett, K.; Briot, D.; Clappier, E.; et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014, 124, 3738–3747.
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208.
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015, 125, 1759–1767.
- Hales, E.C.; Orr, S.M.; Larson Gedman, A.; Taub, J.W.; Matherly, L.H. Notch1 receptor regulates AKT protein activation loop (Thr308) dephosphorylation through modulation of the PP2A phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell acute lymphoblastic leukemia cells. J. Biol. Chem. 2013, 288, 22836–22848.
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660.
- Joshi, I.; Minter, L.M.; Telfer, J.; Demarest, R.M.; Capobianco, A.J.; Aster, J.C.; Sicinski, P.; Fauq, A.; Golde, T.E.; Osborne, B.A. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood 2009, 113, 1689–1698.
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012, 22, 452–465.
- Tremblay, C.S.; Chiu, S.K.; Saw, J.; McCalmont, H.; Litalien, V.; Boyle, J.; Sonderegger, S.E.; Chau, N.; Evans, K.; Cerruti, L.; et al. Small molecule inhibition of Dynamin-dependent endocytosis targets multiple niche signals and impairs leukemia stem cells. Nat. Commun. 2020, 11, 6211.
- Tembhare, P.R.; Sriram, H.; Khanka, T.; Chatterjee, G.; Panda, D.; Ghogale, S.; Badrinath, Y.; Deshpande, N.; Patkar, N.V.; Narula, G.; et al. Flow cytometric evaluation of CD38 expression levels in the newly diagnosed T-cell acute lymphoblastic leukemia and the effect of chemotherapy on its expression in measurable residual disease, refractory disease and relapsed disease: An implication for anti-CD38 immunotherapy. J. Immunother. Cancer 2020, 8, e000630.
- Bride, K.L.; Vincent, T.L.; Im, S.Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018, 131, 995–999.
- Vogiatzi, F.; Winterberg, D.; Lenk, L.; Buchmann, S.; Cario, G.; Schrappe, M.; Peipp, M.; Richter-Pechanska, P.; Kulozik, A.E.; Lentes, J.; et al. Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood 2019, 134, 713–716.
- Ofran, Y.; Ringelstein-Harlev, S.; Slouzkey, I.; Zuckerman, T.; Yehudai-Ofir, D.; Henig, I.; Beyar-Katz, O.; Hayun, M.; Frisch, A. Daratumumab for eradication of minimal residual disease in high-risk advanced relapse of T-cell/CD19/CD22-negative acute lymphoblastic leukemia. Leukemia 2020, 34, 293–295.
- Wang, A.; Song, Z.; Zheng, G.; Nicolazzi, C.; Fromm, J.R.; Shehu, E.; Srinivasan, S.; Chen, X.; Zhu, C.; Blondel, M.C.; et al. Evaluation of Preclinical Activity of Isatuximab in Patients with Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2021, 20, 1916–1925.
- Vora, A.; Bhatla, T.; Teachey, D.; Bautista, F.; Moppett, J.; Velasco Puyó, P.; Micalizzi, C.; Rossig, C.; Shukla, N.; Gilad, G.; et al. Efficacy and Safety of Daratumumab in Pediatric and Young Adult Patients with Relapsed/Refractory T-Cell Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results from Phase 2 DELPHINUS Study. In Proceedings of the 27th Annual Congress of the European Hematology Association, Vienna, Austria, 9–12 June 2022.
- Vakrmanova, B.; Novakova, M.; Riha, P.; Zaliova, M.; Fronkova, E.; Mejstrikova, E.; Stary, J.; Hrusak, O.; Sramkova, L. CD38: A target in relapsed/refractory acute lymphoblastic leukemia-Limitations in treatment and diagnostics. Pediatr. Blood Cancer 2022, 69, e29779.
- Bayon-Calderon, F.; Toribio, M.L.; Gonzalez-Garcia, S. Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 7685.
- Angiolillo, A.L.; Yu, A.L.; Reaman, G.; Ingle, A.M.; Secola, R.; Adamson, P.C. A phase II study of Campath-1H in children with relapsed or refractory acute lymphoblastic leukemia: A Children’s Oncology Group report. Pediatr. Blood Cancer 2009, 53, 978–983.
- Ravandi, F.; Aribi, A.; O’Brien, S.; Faderl, S.; Jones, D.; Ferrajoli, A.; Huang, X.; York, S.; Pierce, S.; Wierda, W.; et al. Phase II study of alemtuzumab in combination with pentostatin in patients with T-cell neoplasms. J. Clin. Oncol. 2009, 27, 5425–5430.
- Dearden, C.E.; Matutes, E.; Catovsky, D. Alemtuzumab in T-cell malignancies. Med. Oncol. 2002, 19, S27–S32.
- Dearden, C. Alemtuzumab in peripheral T-cell malignancies. Cancer Biother. Radiopharm. 2004, 19, 391–398.
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K.; et al. A fully human anti-IL-7Ralpha antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168.
- Hixon, J.A.; Andrews, C.; Kashi, L.; Kohnhorst, C.L.; Senkevitch, E.; Czarra, K.; Barata, J.T.; Li, W.; Schneider, J.P.; Walsh, S.T.R.; et al. New anti-IL-7Ralpha monoclonal antibodies show efficacy against T cell acute lymphoblastic leukemia in pre-clinical models. Leukemia 2020, 34, 35–49.