Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
Hydrogen sulfide (H2S) is an identified and recognized gasotransmitter after nitric oxide and carbon oxide. Atherosclerosis is one of most prevalent cardiovascular diseases worldwide, characterized as chronic inflammation and lipid accumulation in the large arteries. Atherosclerotic plaque rupture or erosion causes the formation of thrombosis or blood clot, resulting in acute events, such as myocardial infraction, or ischemic stroke, contributing to a major mortality rate in the case of human diseases.
- atherosclerosis
- plaque stability
- hydrogen sulfide
1. Introduction
Hydrogen sulfide (H2S) is an identified and recognized gasotransmitter after nitric oxide and carbon oxide. As endogenous methionine catalysis production, H2S major generates from homocysteine trans-sulfide metabolism. Cystathionine β synthase (CBS), cystathionine γ lyase (CSE), cysteine aminotransferase (CAT), and 3-mercaptopyruvate sulfur transferase (3-MST) are major synthetases of H2S. In cardiovascular tissues, CSE and 3-MST are expressed, and CBS to a much lesser extent. There is no accurate experimental evidence of dominant CSE protein expression in cardiovascular tissues. However, in mouse liver (most endogenous H2S generation organ), CSE protein exceeds CBS protein by about 60-fold [1], and CSE mRNA is also about 4.8-fold comparison to CBS and 48.9-fold to 3-MST [2]. Therefore, H2S biosynthetic activity might be sourced from CSE. More and more studies highlight that CSE/H2S protects against cardiovascular diseases, including atherosclerosis in particular.
Atherosclerosis is one of most prevalent cardiovascular diseases worldwide, characterized as chronic inflammation and lipid accumulation in the large arteries. Atherosclerotic plaque rupture or erosion causes the formation of thrombosis or blood clot, resulting in acute events, such as myocardial infraction, or ischemic stroke, contributing to a major mortality rate in the case of human diseases [3]. Although acute coronary syndrome (ACS) or sudden deaths in younger individuals or women patients are usually triggered by plaque erosion, even superficial erosion [4,5], most cardiovascular events occur due to the plaque rupture. Therefore, plaque stability is crucial for preventing cardiovascular events. Some risk factors (such as hyperhomocysteinemia, hypercholesteremia etc.) promote necrotic core formation due to efferocytosis impairment (causing a reduction of apoptotic foam cells clearance), collagen synthesis decrease, and protease (e.g., matrix metallopeptidase) expression/activity elevation, triggering the thickness of fibrotic cap, local acute or chronic inflammation response, intraplaque hemorrhage, and plaque calcification [6], contributing to the pathophysiological modulation of plaque stability.
2. Intraplaque Cell Types and Interactions in Plaque Stability
Plaque stability is mediated by many complicated factors. In 1989, Muller JE and colleagues encapsulated a hypothetic concept of “vulnerable atherosclerotic plaque”, characterized as collagen exposure, or large necrotic core, based on studies of autopsy and angiographic data from myocardial infraction or sudden cardiac death [11]. In 2000, Virmani R. groups showed a “thin” fibrous cap (<65 μm) plaque was “vulnerable” [12]. However, some in vivo human image clinical trials demonstrated that “vulnerable plaque” did not inevitably rupture or cause thrombotic events [13,14,15]. Although these studies are contradictory, they still imply some “vulnerable” characteristics: such as large necrotic core, more macrophages, VSMC apoptosis and necrosis, reduced the collagen level and active inflammation [16].
In human atherosclerotic plaque, approximately 14 different cell populations were identified, including endothelial cells, VSMCs, myeloid cells, and immunocytes (T cells, B cells and mast cells) [17]. Animal and human evidence has demonstrated that each intraplaque cell population plays a critical role in plaque development and stability.
3. Cystathionine γ Lyase/Hydrogen Sulfide Role in Atherosclerotic Plaque Stability
As a gasotransmitter, H2S can easily penetrate the biological membranes due to its lipophilic characteristics. In the body fluid, approximately 1/2 H2S remains undissociated form, and 2/3 exists as HS- ion form at equilibrium with H2S. Recent studies also show that most H2S exists in the body as protein binding form [55]. Thus, binding H2S, HS-, and free H2S maintain a dynamic equilibrium in the body. Once a damage stimulus is received, such as inflammation or reactive oxygen species (ROS), the metabolic activity is increased in cells, and the pH values of local microenvironment are reduced, which can accelerate free H2S generation from HS-. Due to the reduction feature of H2S, the constant consumption caused a decrease of local H2S level, which led to up-regulation of CSE/H2S system (such as acute inflammation [56]) at compensatory phase, or down-regulation of CSE/H2S system at uncompensated period. This may be an interpretation of CSE/H2S reduction in many cardiovascular diseases [56].
More and more research has shown that CSE/H2S play a protective role in atherogenesis [57]. In human and mouse atherosclerotic plaque, CSE protein expression was downregulated [7,58]. Global CSE knockout exacerbated plaque growth by promoting inflammation and oxidative stress [59]. Oppositely, H2S donors attenuated atherogenesis by inhibiting endothelial inflammation and foam cell formation [7,8,9]. For the mechanism, CSE/H2S may modulate VSMC phenotype switch, macrophage polarization, and lipid-phage, endothelia inflammation and monocytes adhesion, and atherothrombosis response, attributed to the genesis and development of plaque [57,60]. Recently, Xiong Q and colleagues showed that H2S enhanced plaque stability by increasing fibrous cap thickness and collagen content, reducing plaque VSMC apoptosis and expression of the collagen-degrading enzyme matrix metallopeptidase-9 (MMP-9) in ApoE knockout mouse model [10]. Here, we discuss the effect of endogenous CSE/H2S in plaque stability, regarding essential risk factors and intraplaque cell population functions.
3.1. H2S Reduced Hypercholesterolemia
Heightening blood cholesterol is a critical process for atherosclerosis initiation and growth. Lowing lipid therapy is the still first-line atherosclerosis therapeutic selection, and it effectively reduces the morbidity and mortality of cardiovascular events. In healthy adult volunteers, circulatory H2S level positively correlated with blood HDL-cholesterol level (r = 0.49), but negatively correlated with LDL/HDL ratio (r = −0.39) [61]. Our previous study also showed that H2S donors lowered serum total cholesterol and LDL cholesterol [9], in part by increasing LDL receptor (LDLR) expression and enhancing LDL uptake activity [62]. In this mechanism, firstly, H2S sulfhydrates Kelch-like ECH-associated protein 1 (Keap-1) at Cys151 site, then promotes nuclear erythroid 2-related factor 2 (Nrf-2) dissociation and nuclear translocation, and up-regulates LDL-related protein 1 (LRP-1) expression in hepatocytes [63]. Secondly, H2S activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)-sterol regulatory element binding proteins 2 (SREBP-2) signaling pathway and inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9), causing LDLR protein elevation [62], which is not dependent on liver X receptor α (LXRα) [64].
3.2. H2S Attenuates Hyperhomocysteinemia
Hyperhomocysteinemia is an independent risk factor for atherosclerosis. Some studies also supported that high blood total homocysteine (Hcy) level correlated with cardiovascular events. However, lowering homocysteine therapy by B type vitamin and/or folic acid by enhancing Hcy re-methylation do not effectively reduce morbidity and mortality of cardiovascular events, but just slightly attenuate stroke mortality for those presenting with hypertension and hyperhomocysteinemia [65]. H2S is a product of homocysteine trans-sulfur catalysis. H2S can increase CSE catalysis Hcy activity or methylenetetrahydrofolate reductase activity by sulfhydration, resulting in the reduction of serum Hcy, then attenuate Hcy-induced macrophages infiltration in the plaque and plaque area [66,67]. In vitro, H2S also reduced Hcy-induced reactive oxygen species (ROS) and endoplasmic reticulum stress [68,69]. These studies also provide a new therapeutic clue using H2S donors, in order to prevent cardiovascular events in patients suffering from hyperhomocysteinemia.
3.3. H2S Modulates VSMC Function in Pathogenesis of Plaque Stability
VSMC function contributes to the formation, growth/development, and even rupture of atherosclerotic plaque [20]. In the arterial wall, endogenous H2S dominant derived from VSMCs [70]. Numerous evidence, direct and indirect, including in vivo and in vitro studies, demonstrated that the endogenous CSE/H2S system may mediate VSMC proliferation, cell death (apoptosis, autophagy, ferroptosis), phage-like differentiation, and calcification, to participate in the pathophysiological process of plaque stability (Figure 1).
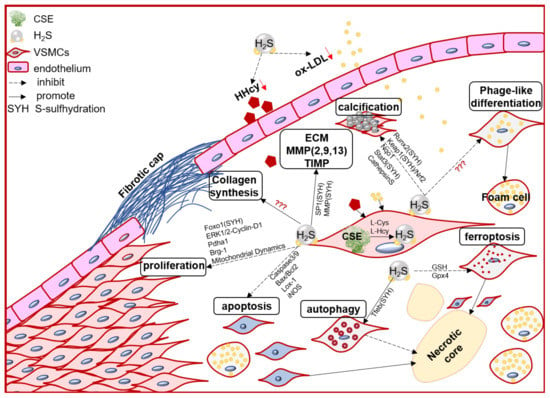
Figure 1. The schematic diagram of endogenous CSE/H2S biological modulation and molecular mechanism in VSMC proliferation, apoptosis, ferroptosis, autophagy, phage-like differentiation, osteochondrogenic conversion/calcification, collagen synthesis/secretion, matrix protease expression and activity, engage in the pathogenesis of atherosclerotic plaque and its stability regulation. Foxo1: forkhead box O1; ERK: Extracellular signal-regulated kinase Stat3: signal transducer and activator of transcription 3; Pdha1: pyruvate dehydrogenase E1 alpha 1; Brg-1: brahma-related gene 1; Lox-1: oxidized low density lipoprotein receptor 1; Runx2: runt related transcription factor 2; Nqo1: NAD(P)H quinone dehydrogenase1; Keap1: Kelch-like ECH-associated protein 1; Nrf2: nuclear factor (erythroid-derived 2)-like 2; GSH: glutathione; Gpx4: glutathione peroxidase 4; Tfeb: transcription factor EB.
3.4. H2S Targets Macrophage Polarization, Foam Cell Formation and Efferocytosis
In the early atherosclerosis stage, local endothelial inflammation promotes monocytes adhesion, then migration into the intimal space. The intimal monocytes proliferate and differentiate into macrophage in initial of atherosclerotic plaque. Plaque macrophages respond to local inflammation and exacerbate the inflammatory cycle by producing proinflammatory cytokines and ECM components, then heightening the retention of lipoprotein to form foam cells [28]. Plaque accumulated cytokines also induce macrophages activation to polarize into pro-inflammation M1 type or anti-inflammation M2 type, promoting (M1) or regressing (M2) plaque growth. Chronic inflammation can promote foam cells apoptosis, lower the efferocytosis of macrophage, resulting in promoting the necrotic core formation and plaque instability [39]. Macrophages also express the CSE/H2S system, which mediates the macrophage polarization, lipid-phage, and efferocytosis to maintain the plaque stability (Figure 2).
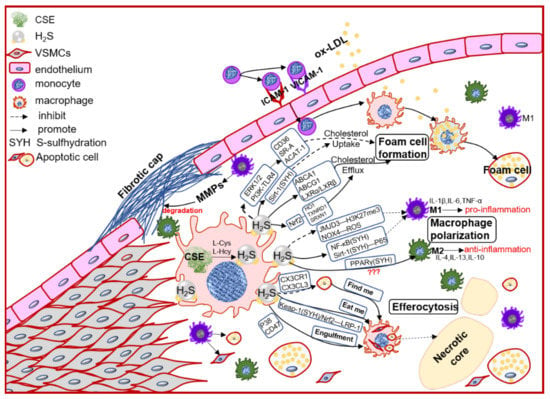
Figure 2. The schematic diagram of CSE/H2S modulation in macrophages polarization, macrophage-derived foam cell formation and efferocytosis, and then participation in the plaque stability. PI3K: phosphatidylinositol 3-kinase; TLR4: toll-like receptor 4; Sirt1: sirtuin 1; ABCA1: ATP-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; SR-A: scavenger receptor A; LXR: liver X receptor; HO1: heme oxygenase 1; TXNRD1: thioredoxin reductase 1; SRXN1: sulfiredoxin-1; JMJD3: jumonji domain-containing protein 3; PPARγ: peroxisome proliferator activated receptor gamma; ACAT-1: acyl-coenzyme A cholesterol acyltransferase-1; NOX4: NADPH oxidase 4; ROS: reactive oxidant species; CX3CR1: chemokine (C-X3-C motif) receptor 1; CX3CL1: chemokine (C-X3-C motif) ligand 1; LRP1: LDL receptor related protein 1; IL-1β: interleukin 1 beta; IL-4: interleukin 4; IL-6: interleukin 6; IL-10: interleukin 10; IL-13: interleukin 13; TNFα: tumor necrosis factor alpha.
3.5. H2S Mediated Adaptive Immunity to Participate into Plaque Stability
Pathological evidence, single-cell RNA sequencing, and cytometry by time of flight (mass cytometry) approaches confirm the presence of T and B cells in human and mouse plaques. In all leukocytes of plaque, approximately 25%–38% are CD3+ cells, and CD4+ T cells approximately 10% [54]. After antigen-presentation, CD4+ T cells differentiated into distinct Th subsets: Th1, Th2, Th17, Treg, T-follicular helper cells (Tfh), and Type 1 regulatory (Tr1) cells [54]. Th1 subset, special expressing T-bet (T-box transcription factor) and secreting IFN-γ, is dominant in human plaques. IFN-γ inhibits VSMC proliferation but increases M1 polarization [134]. Tregs especially express transcription factor FoxP3 (forkhead box P3) and CD25 (IL-2 high affinity receptor), and play a protective role in plaque stability, by releasing IL-10, TGF-β, and inhibiting T-effector cells [135,136]. Therefore, CD4+ T cells regulate local inflammation to affect necrotic core and fibrotic cap formation, contributing to plaque stability.
In primary mouse T lymphocytes and human Jurkat T cells, H2S increases CD69, CD25, and IL-2 expression and promotes T cell proliferation, suggesting T cell activation [137]. In contrast, activated T cells up-regulated CBS and CSE proteins, resulting in increasing H2S production [137]. Interestingly, thrombospondin-1 (CD47 suppressor) mediated T activation is also H2S-dependent [133]. Except for T cell activation, H2S also mediates Tregs proliferation and differentiation. Deletion of CBS [138] or CSE [139] reduced TGF-β-induced Tregs differentiation. By contrast, H2S donors promote Tregs proliferation and differentiation [139]. In the mechanism, H2S sulfhydrated nuclear transcription factor Y subunit beta (NFYB) facilitated methylcytosine dioxygenases Tet1 and Tet2 expression, causing the elevation FoxP3 transcript by its promoter hypomethylation [138]. H2S also activates energy-sensitive AMPK by sulfhydrating live kinase B1 (LKB1) to promote Tregs proliferation and differentiation [139]. H2S heightens local Tregs numbers and IL-10 release, attenuating vascular inflammation and T cell infiltration [139], which may increase plaque stability.
This entry is adapted from the peer-reviewed paper 10.3390/antiox11122356
This entry is offline, you can click here to edit this entry!