LICs, also termed leukemia stem cells (LSCs), were first identified by Dick and his colleagues in studies on acute myeloid leukemia (AML) [
4], characterized by their self-renewal capability and potential to differentiate into leukemic blasts [
5,
6]. Although LICs and LSCs are used interchangeably for AML, their concepts are not necessarily the same [
7]. The LICs more appropriately denote the leukemia cells of origin, whereas the LSCs refer to a distinct subpopulation with the capacity for self-renewal and long-term clonal maintenance at a later stage [
6,
7]. In this context, the LICs are at the apex of the leukemic hierarchy, whereas the LSCs represent cells that can be prospectively isolated from the remainder of the cancer cells based on specific cell surface markers. However, in some cancers such as T-ALL, it is not possible to distinguish LSCs from non-LSCs due to the ill-defined immunophenotypes. In this regard, Dick and others have defined such cells as LICs by their ability to (i) generate leukemia in transplanted xenografts, (ii) self-renew upon serial passages in xenografts, and (iii) give rise to daughter cells with proliferative capacity but that are unable to maintain the tumor clone after serial passages [
6]. Nonetheless, the definition of LICs in T-ALL are not well characterized. In most instances, the T-ALL LICs (T-LICs) and LSCs are still used interchangeably, whereas in some studies, the T-LICs only refer to the transplanted cells with leukemia-initiating capacity. As such, a unified term of T-LICs refers to all cells with leukemia-initiating potential and/or LSC capacity.
2.1. T-LICs in Mouse Models
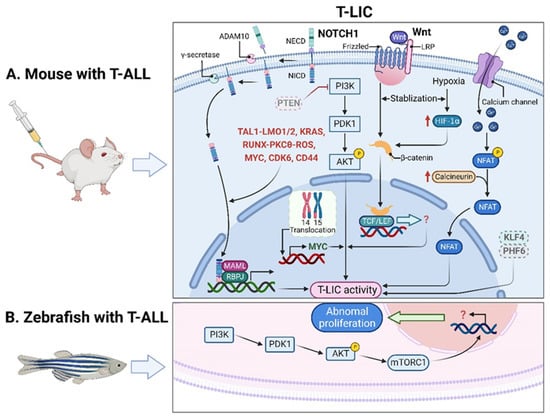
T-LICs were mostly studied in genetically engineered mouse models of T-ALL, among which, the T-cell acute lymphocytic 1 (TAL) basic helix-loop-helix (bHLH) transcription factor 1 (
Tal1)-induced mouse model stands out, since 28% of the
Tal1 transgenic mice develop leukemia [
8]. Additionally, co-expression of
Tal1 with LIM domain only 1 (
Lmo1) or
Lmo2 not only accelerates T-ALL onset and progression [
9,
10] but also provides a favorable context for the acquisition of activating mutations of notch receptor 1 (
NOTCH1) and the emergence of T-LICs [
11]. Thus, the
Tal1-Lmo1/2 transgenic mouse is commonly utilized as a model for characterizing T-LICs [
11,
12]. Moreover, the retroviral or transgenic mouse models of
Notch1-induced T-ALL [
13,
14,
15,
16,
17], KRAS proto-oncogene (
Kras)
G12D-induced T-ALL [
13,
18], and phosphatase and tensin homolog (
Pten)-null T-ALL [
19,
20] are also used for identification of T-LICs in terms of different T-ALL-causing mutations.
Of note, the genes that encode TAL1 and LMO1/2 are recurring targets of chromosomal translocation [
21], and the activating mutations of
NOTCH1 were identified in more than 60% of human T-ALL cases [
22,
23]. Interestingly, NOTCH1 was revealed as a key regulator of human T-LIC activity, since inhibition of the NOTCH1 pathway by γ-secretase inhibitors (GSIs) abolishes T-LIC activity in xenografts and mouse models [
12,
24,
25]. However, the gain-of-function mutations in
NOTCH1 can initiate T-ALL in mouse models but have weak leukemogenic strength, implying that additional cooperating events are required [
13]. Indeed, NOTCH1 modulates T-LIC activity by cooperating with oncogenic TAL1-LMO1/2 transcription factors [
11], KRAS [
13,
18], runt-related transcription factor (RUNX)-mediated regulation of protein kinase C theta (PKCθ) and reactive oxygen species (ROS) [
14], MYC proto-oncogene (MYC) [
16], cyclin dependent kinase 6 (CDK6) [
17], or CD44 [
26] (
Figure 1A).
Figure 1. T-LICs in animal models. (A) The formation and maintenance of T-LICs involve multiple genetic and molecular events, independent or in cooperation with NOTCH1. Briefly, NOTCH1 acts as a ligand-activated transcription factor. The ligand–receptor interaction triggers the cleavage of the NECD by ADAM10 metalloproteases, followed by the subsequent NICD cleavage by γ-secretases. Once released from the membrane, the cytoplasmic NICD translocates into the nucleus, where it activates gene transcription via association with RBPJ DNA-binding protein and MAML transcriptional co-activator. NOTCH1 also modulates T-LIC activity by cooperating with TAL1-LMO1/2, KRAS, RUNX-mediated PKCθ and ROS, MYC, CDK6, or CD44. On the other hand, inactivation of PTEN-induced PI3K-AKT activation, together with the subsequent activation of β-catenin, as well as the aberrant overexpression of MYC induced by t (14;15) translocation, contributes to the onset of T-ALL independent of NOTCH1. Specifically, the binding of Wnt ligand to its receptor Frizzled and/or LRP leads to the activation of Wnt signaling, which stabilizes β-catenin from degradation. Further translocation of β-catenin into the nucleus promotes the downstream gene transcription by binding to TCF/LEF. Finally, the maintenance of T-LIC activity also requires active HIF1α protein, which supports Wnt signaling by promoting accumulation of β-catenin proteins. Activation of calcineurin/NFAT signaling is also critical for T-LIC activity, and the same holds true for the inactivation of KLF4 or PHF6. (B) The increased frequency of T-LICs in zebrafish models might be caused by the abnormal activation of AKT-mTORC1 signaling. ADAM10, a disintegrin and metalloprotease 10; AKT, AKT serine/threonine kinase; CDK6, cyclin dependent kinase 6; HIF1α, hypoxia-induced factor 1 alpha; KLF4, Krüppel-like factor 4; KRAS, KRAS proto-oncogene; LEF, lymphoid enhancer-binding factor; LMO1/2, LIM domain only 1/2; LRP, lipoprotein-receptor-related protein; MAML, mastermind-like; mTORC1, mechanistic target of rapamycin kinase complex 1; MYC, MYC proto-oncogene; NECD, NOTCH extracellular domain; NFAT, nuclear factor of activated T cells; NICD, NOTCH intracellular domain; NOTCH1, notch receptor 1; PHF6, plant homeodomain factor 6; PI3K, phosphatidylinositol 3-kinase; PKCθ, protein kinase C theta; PTEN, phosphatase and tensin homolog; RBPJ, recombination signal binding protein for immunoglobulin kappa J region; ROS, reactive oxygen species; RUNX, runt-related transcription factor; TAL1, T-cell acute lymphocytic 1 (TAL) basic helix-loop-helix (bHLH) transcription factor 1; T-ALL, T-cell acute lymphoblastic leukemia; TCF, T-cell factor; T-LICs, T-ALL leukemia-initiating cells; Wnt, Wingless/Integrated.
2.2. T-LICs in Zebrafish Models
In addition to the studies in mouse xenografts, zebrafish were also employed to recapitulate human T-ALL. Interestingly, leukemic cells from zebrafish models can be transplanted serially, suggesting the presence of T-LICs [
40]. Unlike a minor subpopulation of T-LICs in human and murine T-ALL, the T-LICs are abundant in the
Myc-induced T-ALL zebrafish models based on large-scale single-cell transplantation experiments [
41]. One possible reason might be due to the abnormal activation of AKT-mechanistic target of rapamycin kinase complex 1 (mTORC1) signaling, which enhances the overall frequency of T-LICs in zebrafish [
42] (
Figure 1B).
2.3. Therapies Targeting T-LICs
Currently, the difficulty in the treatment of T-ALL is that the conventional therapeutics mainly target the bulk of leukemic cells but not the T-LICs. In this context, strategies that eradicate T-LICs, the main culprit for relapse, may have significant clinical implications. The above discoveries of T-LICs in animal models pave a new way to target T-LICs by interfering with the pathways that regulate T-LIC activity. For example, the therapies that target NOTCH1 signaling [
12,
24], PI3K-AKT pathway [
20,
43], MYC [
20,
44], calcineurin [
45] or CD44 [
46] can eliminate T-LICs or impair their activities. In addition, the existing therapeutic agents approved for other indications are also identified to eliminate T-LICs. For example, metformin, an anti-diabetic drug, could induce apoptosis of LIC-enriched T-ALL cells, but the mechanism underlying the metformin action remains undetermined [
47]. Parthenolide, an anti-inflammatory agent, can also induce apoptosis of T-LICs, but the individual T-LIC subpopulations within patients have different responses to parthenolide, as disease progression is slowed but not prevented in xenografts from T-ALL patients upon treatment [
48].
3. Leukemic Niches in T-ALL
Many tissues and organs including the BM, thymus, spleen, lymph node (LN) and CNS are involved in the initiation and development of T-ALL. Thus, it is hard to define the “microenvironment” in terms of T-ALL [
49,
50]. Nonetheless, increasing evidence has revealed that the BM, thymic, splenic and CNS niches play key roles in the development of T-ALL [
49,
51,
52,
53,
54,
55]. Reciprocally, T-ALL cells highjack the healthy microenvironment by reshaping it into “pro-leukemic”, thereby promoting malignant progression and chemoresistance while disrupting normal functions [
56,
57,
58,
59,
60,
61].
3.1. Effects of Leukemic Niches on T-ALL Cells
3.1.1. CXC Chemokine Ligand 12 (CXCL12)/CXC Chemokine Receptor 4 (CXCR4) Signaling
CXCL12 (also termed stromal cell derived factor-1, SDF-1) is a chemotactic factor, and its receptor CXCR4 is a highly conserved G-protein-coupled seven transmembrane receptor. Since stromal-vascular BM niches secrete abundant CXCL12, whereas CXCR4 is primarily expressed on HSCs and malignant cells, the CXCL12/CXCR4 axis becomes a key pathway for both normal hematopoiesis and cancer cell homing to the BM [62,63,64,65]. Indeed, T-ALL cells express high surface levels of CXCR4 in a calcineurin-dependent manner, and CXCR4 silencing impairs migration and survival of leukemic cells, as well as T-LIC activity [66]. Likewise, CXCL12 deletion from vascular endothelial niches impedes T-ALL development [67]. Mechanistically, the interaction between CXCL12 and CXCR4 results in activation of many survival pathways including PI3K-AKT and mitogen-activated protein kinase (MAPK) signaling cascades in T-ALL cells [68] (Figure 2A). The CXCL12/CXCR4 signaling also mediates enhanced extramedullary infiltration and dissemination [69,70]. Therefore, the activation of CXCL12/CXCR4 signaling creates a sanctuary microenvironment for T-ALL cells and confers resistance to conventional therapies [68,71]. As such, inhibition of either the CXCL12/CXCR4 interaction or its downstream signaling is of therapeutic benefit in patients with chemoresistant T-ALL [72].
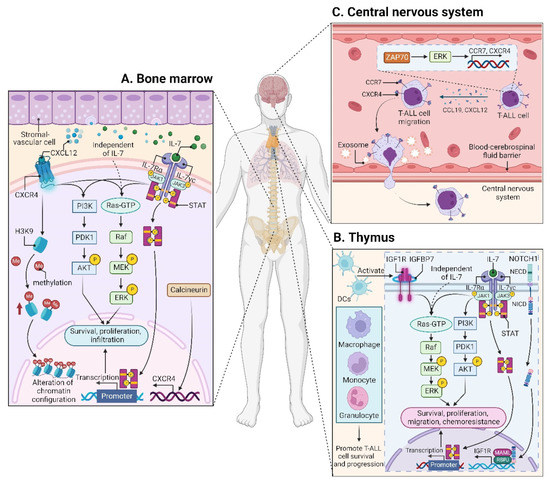
Figure 2. Microenvironments in T-ALL. (
A) The interaction between CXCL12 and CXCR4 within the BM niche leads to activation of many signaling cascades, including PI3K-AKT and MEK-ERK pathways in T-ALL cells. CXCR12 also plays an epigenetic role by promoting H3K9 methylation in T-ALL cells. IL7 is produced by stromal cells in the BM (
A) and thymus (
B), facilitating the activation of JAK-STAT, PI3K-AKT, and MEK-ERK pathways in either an IL7-dependent or IL7-independent manner. (
B) In the thymus, the high levels of IGF1R are supported by the activated NOTCH1 signaling. DCs support T-ALL growth via activating IGF1R and its downstream MAPK signaling; other myeloid cells including macrophages, monocytes, and granulocytes also support T-ALL survival and progression. In addition to IGF1R, IGFBP7, which binds to IGF1R, contributes to T-ALL by prolonging IGF1R activation. (
C) The CCL19/CCR7 signaling is necessary and sufficient for CNS infiltration in T-ALL. Additionally, ZAP70-ERK-induced upregulation of CCR7 and CXCR4, as well as the exosomes isolated from T-ALL cells also confer an increased risk for CNS involvement. BM, bone marrow; CCL19, CC chemokine ligand 19; CCR7, CC chemokine receptor 7; CNS, central nervous system; CXCL12, CXC chemokine ligand 12; CXCR4, CXC chemokine receptor 4; DCs, dendritic cells; ERK, extracellular regulated protein kinase; H3K9, histone 3 lysine 9; IGF1R, insulin-like growth factor 1 receptor; IGFBP7, insulin-like growth factor binding protein 7; IL7, interleukin 7; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; STAT, signal transducer and activator of transcription; ZAP70, zeta-chain-associated protein kinase 70.
3.1.2. Insulin-like Growth Factor 1 (IGF1)/IGF1 Receptor (IGF1R) Signaling
The leukemic niches are highly heterogeneous, being composed of different cell types, extracellular matrix, chemokines, and growth factors. Among them, dendritic cells (DCs) support T-ALL growth via activating IGF1R and its downstream MAPK signaling in the thymus; importantly, the IGF1R signaling is required for DC-mediated T-ALL survival in vitro [
94]. Apart from DCs, myeloid cells, including macrophages, monocytes, and granulocytes, also support T-ALL survival and progression, both in vitro and in vivo; notably, some myeloid cells sensitize T-ALL cells to the IGF1R signaling, indicating that myeloid cells promote T-ALL progression at least in part by activating IGF1R signaling [
95] (
Figure 2B). Nonetheless, it remains unclear whether other signals are required to support T-ALL survival, since monocytes derived from peripheral blood mononuclear cells support T-ALL survival without activating IGF1R [
95].
3.1.3. Interleukin (IL7)/IL7 Receptor (IL7R) Signaling
IL7 is produced by stromal cells in the BM and thymus. It binds to IL7R, a heterodimer composed of an IL7Rα chain and a γ
c chain [
101]. The IL7/IL7R signaling is critical to the survival and proliferation of thymocytes, and the tight regulation of IL7 and IL7Rα is essential for T-cell development. Hence, it is not surprising that aberrant expression or dysfunction of the IL7/IL7R axis contributes to the pathogenesis of T-ALL [
102,
103]. Several lines of evidence have demonstrated that the somatic gain-of-function mutation in
IL7Ra exon 6 is a known driver of T-ALL, occurring in roughly 9% of pediatric and 12% of adult T-ALL cases [
104,
105,
106,
107,
108,
109]. Mechanistically,
IL7Ra mutations induce activation of Janus kinase (JAK)-signal transducer and activator of transcription (STAT), PI3K-AKT, and MAPK kinase (MEK)-extracellular regulated protein kinase (ERK) pathways [
76,
104,
110]. Overexpression of wild-type IL7Rα also promotes T-cell tumorigenesis via JAK-STAT, PI3K-AKT, and cell-cycle-related signaling, even in the absence of
IL7Rα mutational activation [
111]. Moreover, high levels of IL7Rα expression lead to increased T-LIC activity mediated by the activating mutations in
NOTCH1 [
112] or the loss-of-function mutations in
DYNAMIN2 [
113]. These findings pinpoint that not only mutational activation of
IL7Rα but also high IL7Rα levels are oncogenic in T-ALL (
Figure 2B). In addition to IL7Rα, mutations in other components of the IL7R-mediated signaling cascade (e.g., JAK1, JAK3, STAT5) were also identified as critical drivers for T-ALL [
106,
108,
114,
115,
116,
117]. These mutations and the resulting aberrant signaling provide a therapeutic window of opportunity [
108].
3.1.4. CC Chemokine Ligand 19 (CCL19)/CC Chemokine Receptor 7 (CCR7) Signaling
Patients with T-ALL are at a high risk of CNS relapse [
21], which requires intensified intrathecal chemotherapy and systemic administration of CNS-penetrating therapeutics. Unfortunately, the mechanisms accounting for the infiltration of T-ALL cells into CNS remain incompletely understood, and little is known about their crosstalk.
The CCL19/CCR7 signaling was identified as a key CNS entry signal, which is both necessary and sufficient for T-ALL cells targeting the CNS. Silencing either CCR7 or CCL19 specifically inhibits CNS infiltration [
118]. Furthermore, the BM infiltration constitutes a prerequisite for CNS pathology in T-ALL via the CXCR4-mediated signaling [
70]. In this sense, CNS invasion by T-ALL cells is very likely attributed to the BM microenvironmental alterations. Interestingly, both CCR7 and CXCR4 are upregulated by zeta-chain-associated protein kinase 70 (ZAP70) via the activation of ERK signaling, and high expression of ZAP70-CCR7 confers an increased risk for CNS involvement in T-ALL patients [
119] (
Figure 2C). Therefore, therapeutic monoclonal antibodies (mAbs) targeting CCR7 not only display a strong in vitro complement-dependent cytotoxicity (CDC) and an in vivo anti-tumor activity, but also show efficacy in eradicating leukemic cells from LN and CNS [
120]. In addition to the CCR7-mediated CNS infiltration, the exosomes isolated from a T-ALL cell line P12 but not the B-cell acute lymphoblastic leukemia (B-ALL) cell lines facilitate CNS invasion across the blood–cerebrospinal fluid (CSF) barrier without disrupting the barrier integrity [
121] (
Figure 2C), highlighting the contribution of T-ALL-derived exosomes to CNS infiltration, although further studies are needed to clarify the molecular mechanisms.
3.2. Microenvironmental Alterations
3.2.1. BM Microenvironment
Modification of the BM microenvironment by leukemia cells was well-characterized in B-ALL and AML [126,127,128], whereas the research on the alterations of the BM microenvironment by T-ALL cells is very limited. In 2016, an elegant work reported that an accumulated T-ALL burden within the BM leads to rapid and selective remodeling of the endosteal space, resulting in a complete loss of mature osteoblasts and impairment of normal HSCs [56].
3.2.2. Thymic Microenvironment
The thymus is a conserved primary lymphoid organ where progenitors from the BM commit to T-cell lineage development. Thus, thymocytes were long thought to be short-lived cells with no self-renewal capacity. However, two independent groups have updated the notion with the fact that the thymus is capable of sustaining T-cell development and export independently from BM contribution, a state termed as thymus autonomy [
129,
130]. However, thymus autonomy must be tightly regulated, as prolonged autonomy allows profound alterations in the thymic microenvironment, contributing to the initiation and propagation of T-ALL [
131]. In this regard, T-ALL is proposed as a consequence of thymus autonomy [
132].
3.2.3. Splenic Microenvironment
The spleen is a common extramedullary site of leukemia, and splenomegaly is associated with poor clinical outcome in many subtypes of leukemias including T-ALL [
133,
134,
135]. However, the spleen was largely ignored as a tumor microenvironment site due to the difficulties in obtaining biopsy samples from patients. In a previous work, the leukemia-associated macrophages within the spleen were revealed to recruit T-ALL cells potently and stimulate their proliferation [
53]. Further investigation demonstrated that the mice transplanted with the spleen-resident T-ALL cells exhibit a short life span compared to those transplanted with T-ALL cells from the BM, suggesting an increased potency in T-ALL cells induced by the splenic microenvironment [
54]. There is even evidence that splenectomies either before or after the injection of T-ALL cells prolong the survival of mice but do not inhibit the development of T-ALL [
54]. In contrast, removal of the spleen in a genetic mouse model of delta-like canonical Notch ligand 4 (DLL4)-driven T-ALL fully protects against leukemia development [
55], indicating a crucial role of the spleen in DLL4-driven T-ALL.
4. Preclinically- and Clinically-Evaluated Precision Medicine for T-ALL
4.1. Agents Targeting Aberrant Pathways
4.1.1. NOTCH1 Signaling
NOTCH1 signaling is an attractive therapeutic target for T-ALL due to its essential roles in T-ALL initiation and progression. Therefore, its inhibition via GSIs was investigated as a potential targeted therapeutic in preclinical studies [
138], followed by a series of clinical trials [
78,
79]. Despite a hint of clinical efficacy, GSIs were not applied to clinical practice. One major reason is that GSIs have activity against T-ALL with
NOTCH1 mutations but not those with
PTEN deficiency and activation of PI3K-AKT signaling [
30], as well as constitutive MYC expression [
139]. Another key problem is the occurrence of gastrointestinal toxicity and unsatisfactory clinical trials caused by the use of broad-spectrum GSIs [
79,
140,
141]. In this context, selective targeting of the certain components of γ-secretase complexes [
142] or putative responders [
139] may improve antileukemic efficacy while sparing patients from excessive toxicities.
4.1.2. BCL2 Signaling
BCL2, a key regulator of the apoptotic pathway, has emerged as another attractive molecular target in T-ALL due to its high expression in T-ALL cells [
146]. Navitoclax (ABT-263) is the first generation of the BCL2 inhibitor, whereas venetoclax (ABT-199) is a selective BCL2 inhibitor [
147]. The antileukemic effects of venetoclax in T-ALL, either as a monotherapy or in combination with chemotherapy, were evaluated experimentally [
146] and clinically.
4.1.3. JAK-STAT Signaling
Active signaling via JAK1/2 is widely reported in T-ALL cells and leukemic niches, making JAK1/2 inhibitor ruxolitinib a promising agent. However, in preclinical PDX models of T-ALL, treatment with ruxolitinib as a single agent exhibits dramatic efficacy but fails to achieve CR [
153].
4.1.4. PI3K-AKT-mTOR Signaling
Inhibitors that target PI3K-AKT-mTOR pathway were largely evaluated under clinical trials for T-ALL. However, the complex interplay between NOTCH1-PI3K-AKT and PTEN-PI3K-AKT signaling makes the targeted therapy much trickier. Treatment with GSIs in PTEN-deficient cells results in hyperactivation of AKT [
158]. Dual inhibition of PI3K and mTOR pathways triggers NOTCH1-MYC activity [
159]. These discoveries highlight the necessity of combination therapies by concurrently blocking different pathways or a common downstream effector in T-ALL.
4.1.5. CDK4/6-Mediated Signaling
Both CDK4 and CDK6 are the targets of NOTCH1 signaling and contribute to the deregulated cell-cycle progression in T-ALL cells [
161]. Thus, inhibition of CDK4/6 activity efficiently suppresses T-ALL progression in vivo but most likely not target T-LICs, because interruption of drug administration leads to disease relapse [
162].
4.1.6. Other Signaling Pathways
One recent study has identified a novel small-molecule inhibitor Dynole 34-2, which is a specific and potent inhibitor of Dynamin. Dynole 34-2 not only impairs T-LIC activity but also sensitizes them to chemotherapy. More essentially, Dynole 34-2 exhibits efficacy against multiple niche signals in T-LICs including IL-7, NOTCH1, etc. [
35]. This discovery provides a significant advance in developing therapeutic strategies by targeting T-LICs concurrently with multiple microenvironmental signals.
4.2. Antibody-Based Therapy
4.2.1. CD38 mAbs
T cells can be activated via T cell receptor or by triggering multiple cell surface molecules including CD38. Although it is unknown whether T-LICs are positive or negative for CD38, blasts from patients with T-ALL have robust surface expression of CD38 at the time of diagnosis, 1 month post induction, and relapse, making it an ideal target for T-ALL patients who relapse or do not respond to conventional chemotherapies [
171,
172]. Daratumumab, a fully human mAb against CD38, was identified to be highly effective in T-ALL PDX models (14 out of 15), and the only PDX model that failed to respond to daratumumab showed low expression of CD38 [
172]. More essentially, daratumumab can effectively eradicate minimal residual disease in preclinical models of pediatric T-ALL and high-risk advanced relapse T-ALL [
173,
174], providing compelling evidence for the potential clinical efficacy of daratumumab in T-ALL. As such, clinical trials testing the efficacy of daratumumab in T-ALL are currently being evaluated, and the same is true for another anti-CD38 mAb, isatuximab [
83]. Very recently, the preliminary results released from a phase 2 trial of daratumumab in combination with chemotherapy (NCT03384654) are encouraging, with an overall response rate of 83.3% in children and 60% in young adults with R/R T-ALL [
175]. In spite of this, a recent clinical report indicated that not all patients with R/R T-ALL responded to daratumumab administration [
176]. One major reason is the loss of CD38, which could be overcome by monitoring CD38 during treatment or using other antibodies by targeting different epitopes or molecules.
4.2.2. CD52 mAbs
CD52 is widely expressed on normal and malignant B and T cells, and its function remains largely unknown. Alemtuzumab is a humanized anti-CD52 mAb that causes cell death by antibody-dependent cell mediated cytotoxicity (ADCC), CDC, and apoptosis upon binding to CD52 [
178]. Although different clinical trials have tested the efficacy of alemtuzumab for T-cell malignancies including T-ALL [
84,
85,
179,
180], the activity of single-agent alemtuzumab is limited in children with R/R T-ALL based on a Phase 2 study (NCT00089349) [
84]. Additionally, no response was observed in adults with relapsed T-ALL when combining alemtuzumab with pentostatin (NCT00453193) [
85]. As a consequence, no new trials targeting CD52 have been initiated for T-ALL.
4.2.3. IL7Rα mAbs
Apart from the inhibitors of IL7R-mediated signaling as aforementioned, a more direct strategy is to explore antibody-based treatment by targeting IL7Rα itself. Notably, a fully human anti-IL7Rα antibody (B12) that recognizes both the wild-type form and different gain-of-function mutated variants was generated using combinatorial phage-display libraries and antibody reformatting [
181]. B12 not only promotes T-ALL cell death in vitro and delays T-ALL development in vivo, but it also sensitizes T-ALL cells to dexamethasone. More importantly, B12 exhibits a remarkably fast internalization with substantial trafficking into lysosomes, making it an ideal deliverer for toxin conjugates. Recently, another two new chimeric mAbs against human IL7Rα (4A10 and 2B8) that target non-overlapping IL7Rα epitopes were reported [
182]. Both 4A10 and 2B8 mediate increased ADCC against patient-derived T-ALL cells and lead to effective anti-leukemia responses in vivo. Unlike B12, 4A10 and 2B8 cannot induce rapid internalization, but this feature would be desirable for promoting ADCC. Although anti-IL7Rα mAbs were only investigated in preclinical models, future clinical evaluation in T-ALL is eagerly anticipated.