Cardiac remodeling is defined as a group of molecular, cellular, and interstitial changes that clinically manifest as changes in the heart’s size, mass, geometry, and function after different stimuli. It is important to emphasize that remodeling plays a pathophysiological role in the onset and progression of ventricular dysfunction and subsequent heart failure. Therefore, strategies to mitigate this process are critical. Different factors, including neurohormonal activation, can regulate the remodeling process and increase cell death, alterations in contractile and regulatory proteins, alterations in energy metabolism, changes in genomics, inflammation, changes in calcium transit, metalloproteases activation, fibrosis, alterations in matricellular proteins, and changes in left ventricular geometry, among other mechanisms.
- antioxidants
- heart failure
- cardiac remodeling
1. Introduction
2. Cardiac Remodeling
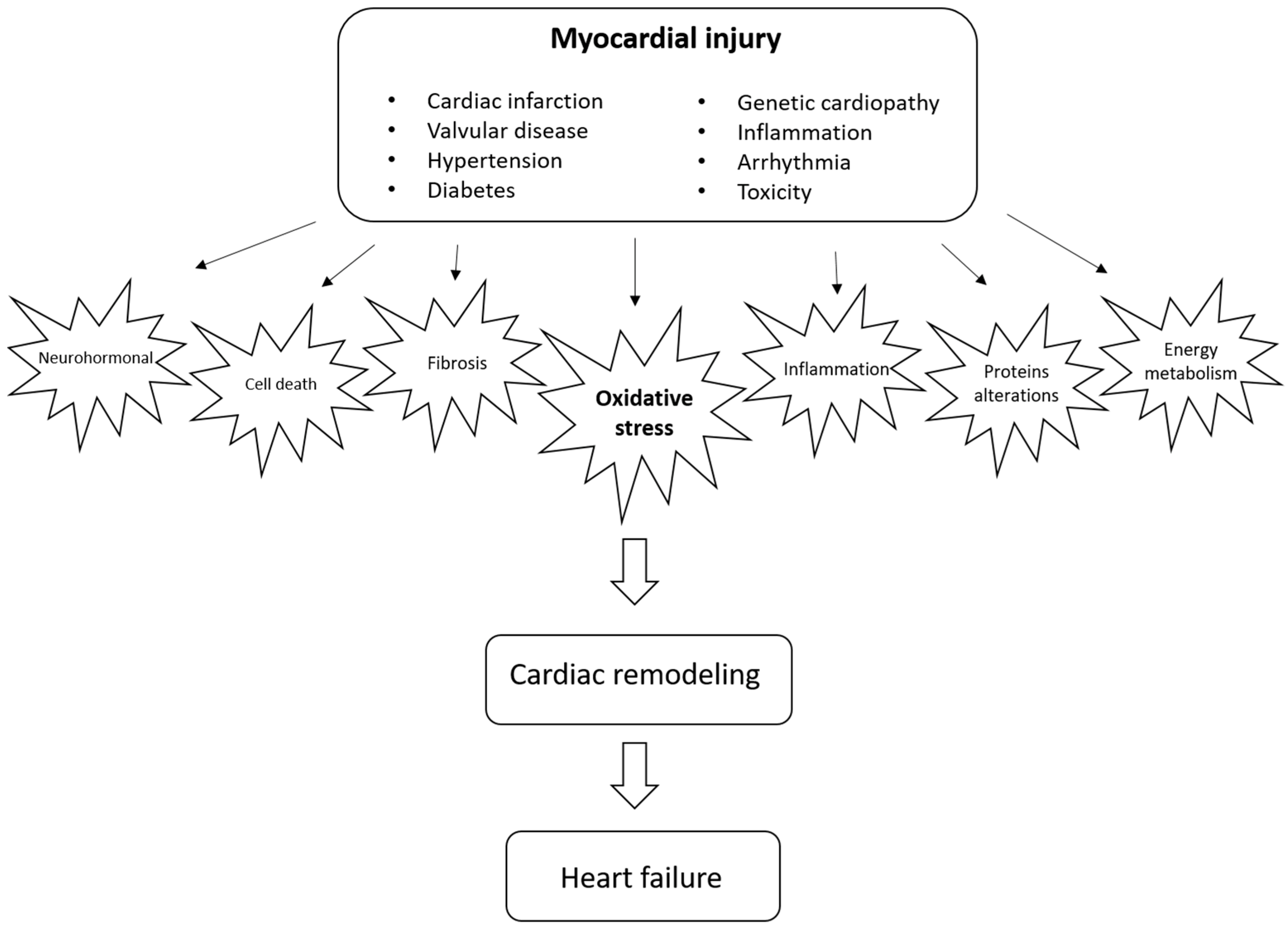
3. Oxidative Stress as a Potential Modulator of Cardiac Remodeling
This entry is adapted from the peer-reviewed paper 10.3390/antiox11122371
References
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac remodeling: Concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq. Bras. Cardiol. 2016, 106, 62–69.
- Yang, D.; Liu, H.Q.; Liu, F.Y.; Tang, N.; Guo, Z.; Ma, S.Q.; An, P.; Wang, M.Y.; Wu, H.M.; Yang, Z.; et al. The roles of noncardiomyocytes in cardiac remodeling. Int. J. Biol. Sci. 2020, 16, 2414–2429.
- Leancă, S.A.; Crișu, D.; Petriș, A.O.; Afrăsânie, I.; Genes, A.; Costache, A.D.; Tesloianu, D.N.; Costache, I.I. Left ventricular remodeling after myocardial infarction: From physiopathology to treatment. Life 2022, 12, 1111.
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931.
- Ramachandra, C.J.A.; Cong, S.; Chan, X.; Yap, E.P.; Yu, F.; Hausenloy, D.J. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic. Biol. Med. 2021, 166, 297–312.
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190.
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74.
- Halliwell, B.; Gutteridge, J.M.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 119, 598–620.
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33.
- López-Alarcón, C.; Denicola, A. Evaluating the antioxidant capacity of natural products: A review on chemical and cellular-based assays. Anal. Chim. Acta 2013, 763, 1–10.
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress–activated signaling pathways mediators of insulin resistance and β-Cell dysfunction? Diabetes 2003, 52, 1–8.
- Gutteridge, J.M.C. Biological origin of free radicals, and mechanisms of antioxidant protection. Chem. Biol. Interact. 1994, 91, 133–140.
- Hou, Y.C.; Janczuk, A.; Wang, P.G. Current trends in the development of nitric oxide donors. Curr. Pharm. Des. 1999, 5, 417–441.
- Goldblum, R.R.; McClellan, M.; White, K.; Gonzalez, S.J.; Thompson, B.R.; Vang, H.X.; Cohen, H.; Higgins, L.; Markowski, T.W.; Yang, T.Y.; et al. Oxidative stress pathogenically remodels the cardiac myocyte cytoskeleton via structural alterations to the microtubule lattice. Dev. Cell 2021, 56, 2252–2266.
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090.
- Sabri, A.; Hughie, H.H.; Lucchesi, P.A. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid. Redox Signal. 2003, 5, 731–740.
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912.
- Hayashidani, S.; Tsutsui, H.; Ikeuchi, M.; Shiomi, T.; Matsusaka, H.; Kubota, T.; Iamanaka-Yoshida, K.; Itoh, T.; Takeshita, A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1229–H1235.
- Stones, R.; Benoist, D.; Peckham, M.; White, E. Microtubule proliferation in right ventricular myocytes of rats with monocrotaline-induced pulmonary hypertension. J. Mol. Cell Cardiol. 2013, 56, 91–96.
- Ohi, R.; Zanic, M. Ahead of the curve: New insights into microtubule dynamics. F1000Research 2016, 5, 314.
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435.
- Ogawa, H.; Kurebayashi, N.; Yamazawa, T.; Murayama, T. Regulatory mechanisms of ryanodine receptor/Ca2+ release channel revealed by recent advancements in structural studies. J. Muscle Res. Cell Motil. 2021, 42, 291–304.
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, C1–C24.
- Kawakami, M.; Okabe, E. Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol. Pharmacol. 1998, 53, 497–503.
- Spinale, F.G. Bioactive peptide signaling within the myocardial interstitium and the matrix metalloproteinases. Circ. Res. 2002, 91, 1082–1084.
- Mitra, A.; Datta, R.; Rana, S.; Sarkar, S. Modulation of NFKB1/p50 by ROS leads to impaired ATP production during MI compared to cardiac hypertrophy. J. Cell Biochem. 2018, 119, 1575–1590.
- Kang, D.; Hamasaki, N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann. N. Y. Acad. Sci. 2005, 1042, 101–108.
- Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Matic, M.; Simic, D.; Radovanovic, S.; Simic, T. Novel biomarkers of heart failure. Adv. Clin. Chem. 2017, 79, 93–152.
- Tang, W.H.W.; Tong, W.; Troughton, R.W.; Martin, M.G.; Shrestha, K.; Borowski, A.; Jasper, S.; Hazen, S.L.; Klein, A.L. Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. J. Am. Coll. Cardiol. 2007, 49, 2364–2370.
- Tromp, J.; Khan, M.A.F.; Mentz, R.J.; O’Connor, C.M.; Metra, M.; Dittrich, H.C.; Ponikowski, P.; Teeerlink, J.R.; Cotteer, G.; Davison, B.; et al. Biomarker profiles of acute heart failure patients with a mid-range ejection fraction. JACC Heart Fail. 2017, 5, 507–517.
- Wang, H.; Chen, Q.; Li, Y.; Jing, X.; Yang, J. Prognostic value of growth differentiation factor-15 in Chinese patients with heart failure: A prospective observational study. Cardiol. J. 2018, 25, 245–253.
- Yamagami, F.; Tajiri, K.; Doki, K.; Hattori, M.; Honda, J.; Aita, S.; Harunari, T.; Yamasaki, H.; Murakoshi, N.; Sekiguchi, Y.; et al. Indoxyl sulphate is associated with atrial fibrillation recurrence after catheter ablation. Sci. Rep. 2018, 8, 17276.
- Wollert, K.C.; Kempf, T. Growth differentiation factor 15 in heart failure: An update. Curr. Heart Fail. Rep. 2012, 9, 337–345.
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308.
- Yang, P.S.; Kim, T.; Uhm, J.S.; Park, S.; Joung, B.; Lee, M.H.; Pak, H.N. High plasma level of soluble RAGE is independently associated with a low recurrence of atrial fibrillation after catheter ablation in diabetic patient. Europace 2016, 18, 1711–1718.
- Goidescu, C.M.; Chiorescu, R.M.; Diana, M.H.L.; Mocan, M.; Stoia, M.A.; Anton, F.P.; Farcas, A.D. ACE2 and Apelin-13: Biomarkers with a prognostic value in congestive heart failure. Dis. Markers 2021, 2021, 5569410.