The brain is highly sensitive to oxidative stress due to its high oxygen consumption, abundance of unsaturated fatty acids which are prone to oxidation, and low antioxidant levels. It is a metabolically active and a high energy demanding organ that relies heavily on mitochondria for its energy needs. Majority of oxygen consumed by mitochondria during oxidative phosphorylation is coupled to ATP synthesis while ~4% contributes to the generation of superoxides which are further metabolized to reactive oxygen species (ROS). ROS modify proteins causing functional and structural damage to biomolecules. Prolonged exposure to reactive oxygen species (ROS) also damages DNA, mitochondrial membranes, and lipids, impairing its metabolic functions including synthesis of ATP, fatty acid oxidation and metabolism of essential biomolecules.
- oxidative stress
- glutathione
- glutaredoxin
- protein thiol
- ischemia
1. Brain Glutathione
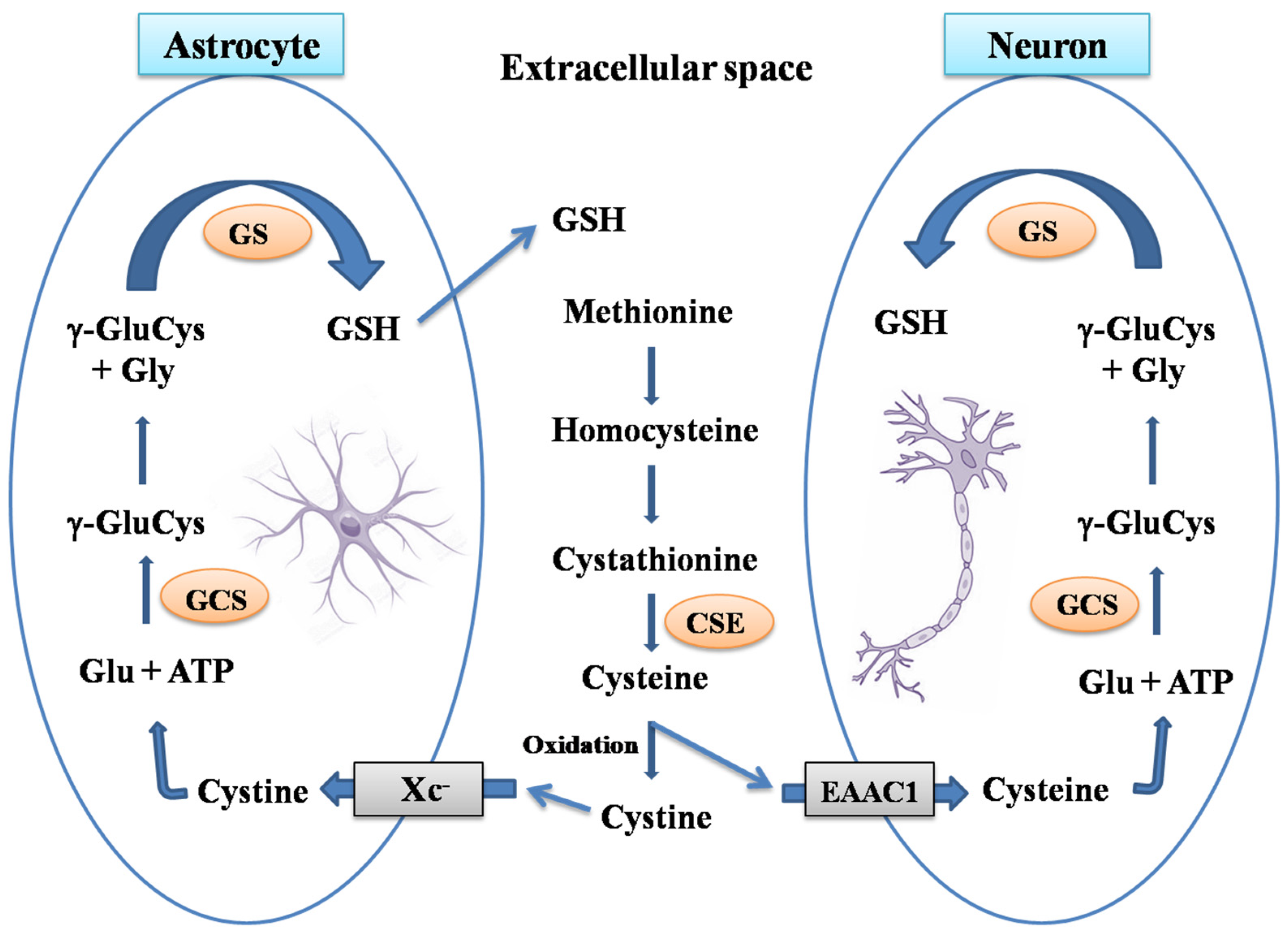
2. Glutaredoxin
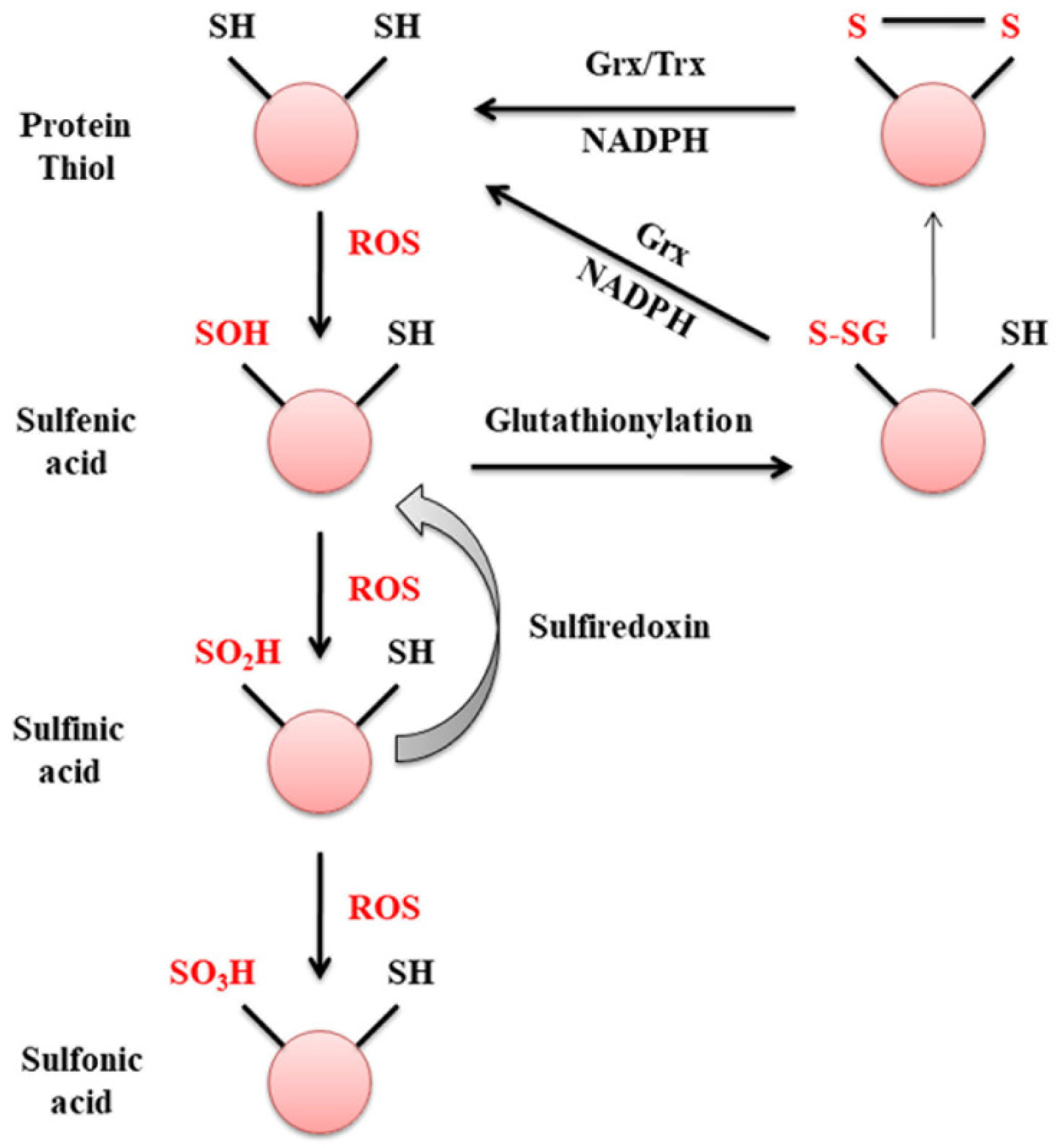
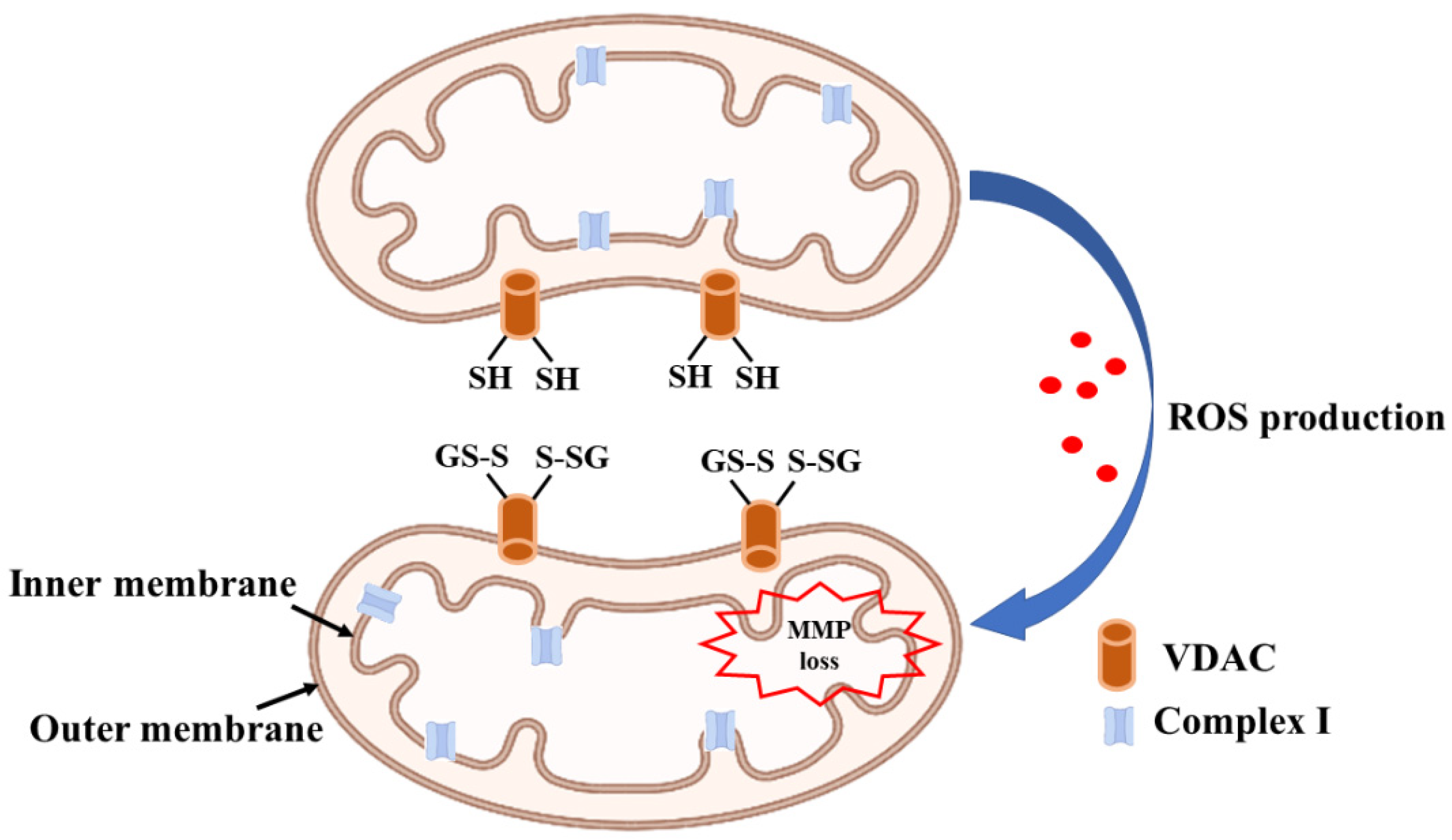
3. Glutaredoxin and Mitochondrial Dysfunction
4. Excitotoxicity and Glutaredoxin
5. Ischemic Reperfusion Injury
This entry is adapted from the peer-reviewed paper 10.3390/antiox11122334
References
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758.
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59.
- Vali, S.; Mythri, R.B.; Jagatha, B.; Padiadpu, J.; Ramanujan, K.S.; Andersen, J.K.; Gorin, F.; Bharath, M.M.S. Integrating Glutathione Metabolism and Mitochondrial Dysfunction with Implications for Parkinson’s Disease: A Dynamic Model. Neuroscience 2007, 149, 917–930.
- Wang, X.F.; Cynader, M.S. Astrocytes Provide Cysteine to Neurons by Releasing Glutathione. J. Neurochem. 2000, 74, 1434–1442.
- Aoyama, K.; Watabe, M.; Nakaki, T. Modulation of Neuronal Glutathione Synthesis by EAAC1 and Its Interacting Protein GTRAP3-18. Amino Acids 2012, 42, 163–169.
- Diwakar, L.; Ravindranath, V. Inhibition of Cystathionine-γ-Lyase Leads to Loss of Glutathione and Aggravation of Mitochondrial Dysfunction Mediated by Excitatory Amino Acid in the CNS. Neurochem. Int. 2007, 50, 418–426.
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440.
- Braidy, N.; Zarka, M.; Jugder, B.E.; Welch, J.; Jayasena, T.; Chan, D.K.Y.; Sachdev, P.; Bridge, W. The Precursor to Glutathione (GSH), γ-Glutamylcysteine (GGC), Can Ameliorate Oxidative Damage and Neuroinflammation Induced by Aβ40 Oligomers in Human Astrocytes. Front. Aging Neurosci. 2019, 10, 177.
- Skvarc, D.R.; Dean, O.M.; Byrne, L.K.; Gray, L.; Lane, S.; Lewis, M.; Fernandes, B.S.; Berk, M.; Marriott, A. The Effect of N-Acetylcysteine (NAC) on Human Cognition—A Systematic Review. Neurosci. Biobehav. Rev. 2017, 78, 44–56.
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-Glutamylcysteine (γ-GC) Lessens Oxidative Stress, Brain Inflammation and Amyloid Pathology and Improves Spatial Memory in a Murine Model of AD. Neurochem. Int. 2021, 144, 104931.
- Kinoshita, C.; Kikuchi-Utsumi, K.; Aoyama, K.; Suzuki, R.; Okamoto, Y.; Matsumura, N.; Omata, D.; Maruyama, K.; Nakaki, T. Inhibition of MiR-96-5p in the Mouse Brain Increases Glutathione Levels by Altering NOVA1 Expression. Commun. Biol. 2021, 4, 1–12.
- Gorelenkova Miller, O.; Mieyal, J.J. Critical Roles of Glutaredoxin in Brain Cells—Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2019, 30, 1352–1368.
- Shelton, D.M.; Chock, P.B.; Mieyal, J.J. S-Glutathionylation As a Mechanism of Redox Signal Transduction and Regulation of Protein Translocation, and the Central Role of Glutaredoxin. Antioxid. Redox Signal. 2005, 7, 348–367.
- Van Bergen, L.A.H.; Roos, G.; De Proft, F. From Thiol to Sulfonic Acid: Modeling the Oxidation Pathway of Protein Thiols by Hydrogen Peroxide. J. Phys. Chem. A 2014, 118, 6078–6084.
- Mieyal, J.J.; Starke, D.W.; Chung, J.S.; Gravina, S.A.; Dothey, C. Thioltransferase in Human Red Blood Cells: Purification and Properties. Biochemistry 1991, 30, 6088–6097.
- Chrestensen, C.A.; Eckman, C.B.; Starke, D.W.; Mieyal, J.J. Cloning, Expression and Characterization of Human Thioltransferase (Glutaredoxin) in E. coli. FEBS Lett. 1995, 374, 25–28.
- Balijepalli, S.; Tirumalai, P.S.; Swamy, K.V.; Boyd, M.R.; Mieyal, J.J.; Ravindranath, V. Rat Brain Thioltransferase: Regional Distribution, Immunological Characterization, and Localization by Fluorescent in Situ Hybridization. J. Neurochem. 1999, 72, 1170–1178.
- Balijepalli, S.; Boyd, M.R.; Ravindranath, V. Human Brain Thioltransferase: Constitutive Expression and Localization by Fluorescence in Situ Hybridization. Mol. Brain Res. 2000, 85, 123–132.
- Griffith, O.W.; Meister, A. Origin and Turnover of Mitochondrial Glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672.
- Ravindranath, V.; Reed, D.J. Glutathione Depletion and Formation of Glutathione-Protein Mixed Disulfide Following Exposure of Brain Mitochondria to Oxidative Stress. Biochem. Biophys. Res. Commun. 1990, 169.
- Olafsdottir, K.; Reed, D.J. Retention of Oxidized Glutathione by Isolated Rat Liver Mitochondria during Hydroperoxide Treatment. BBA Gen. Subj. 1988, 964, 377–382.
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible Glutathionylation of Complex I Increases Mitochondrial Superoxide Formation. J. Biol. Chem. 2003, 278, 19603–19610.
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 Catalyzes the Reversible Oxidation and Glutathionylation of Mitochondrial Membrane Thiol Proteins: Implications for Mitochondrial Redox Regulation and Antioxidant Defense. J. Biol. Chem. 2004, 279, 47939–47951.
- Kenchappa, R.S.; Ravindranath, V. Glutaredoxin Is Essential for Maintenance of Brain Mitochondrial Complex I: Studies with MPTP. FASEB J. 2003, 17, 717–719.
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and Expression of a Novel Human Glutaredoxin (Grx2) with Mitochondrial and Nuclear Isoforms. J. Biol. Chem. 2001, 276, 26269–26275.
- Lönn, M.E.; Hudemann, C.; Berndt, C.; Cherkasov, V.; Capani, F.; Holmgren, A.; Lillig, C.H. Expression Pattern of Human Glutaredoxin 2 Isoforms: Identification and Characterization of Two Testis/Cancer Cell-Specific Isoforms. Antioxid. Redox Signal 2008, 10, 547–557.
- Karunakaran, S.; Saeed, U.; Ramakrishnan, S.; Koumar, R.C.; Ravindranath, V. Constitutive Expression and Functional Characterization of Mitochondrial Glutaredoxin (Grx2) in Mouse and Human Brain. Brain Res. 2007, 1185.
- Enoksson, M.; Fernandes, A.P.; Prast, S.; Lillig, C.H.; Holmgren, A.; Orrenius, S. Overexpression of Glutaredoxin 2 Attenuates Apoptosis by Preventing Cytochrome c Release. Biochem. Biophys. Res. Commun. 2005, 327, 774–779.
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carrì, M.T. Glutaredoxin 2 Prevents Aggregation of Mutant SOD1 in Mitochondria and Abolishes Its Toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542.
- Petronilli, V.; Costantini, P.; Scorrano, L.; Colonna, R.; Passamonti, S.; Bernardi, P. The Voltage Sensor of the Mitochondrial Permeability Transition Pore Is Tuned by the Oxidation-Reduction State of Vicinal Thiols. Increase of the Gating Potential by Oxidants and Its Reversal by Reducing Agents. J. Biol. Chem. 1994, 269, 16638–16642.
- Saeed, U.; Durgadoss, L.; Valli, R.K.; Joshi, D.C.; Joshi, P.G.; Ravindranath, V. Knockdown of Cytosolic Glutaredoxin 1 Leads to Loss of Mitochondrial Membrane Potential: Implication in Neurodegenerative Diseases. PLoS ONE 2008, 3, e2459.
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial Membrane Potential Regulates Matrix Configuration and Cytochrome c Release during Apoptosis. Cell Death Differ. 2003, 10, 709–717.
- Ravindranath, V. Neurolathyrism: Mitochondrial Dysfunction in Excitotoxicity Mediated by L-β-Oxalyl Aminoalanine. Neurochem. Int. 2002, 40, 505–509.
- Sadashiva Pai, K.; Ravindranath, V. L-BOAA Induces Selective Inhibition of Brain Mitochondrial Enzyme, NADH-Dehydrogenase. Brain Res. 1993, 621, 215–221.
- Sriram, K.; Shankar, S.K.; Boyd, M.R.; Ravindranath, V. Thiol Oxidation and Loss of Mitochondrial Complex I Precede Excitatory Amino Acid-Mediated Neurodegeneration. J. Neurosci. 1998, 18, 10287–10296.
- Kenchappa, R.S.; Diwakar, L.; Boyd, M.R.; Ravindranath, V. Thioltransferase (Glutaredoxin) Mediates Recovery of Motor Neurons from Excitotoxic Mitochondrial Injury. J. Neurosci. 2002, 22, 8402–8410.
- Diwakar, L.; Kenchappa, R.S.; Annepu, J.; Ravindranath, V. Downregulation of Glutaredoxin but Not Glutathione Loss Leads to Mitochondrial Dysfunction in Female Mice CNS: Implications in Excitotoxicity. Neurochem. Int. 2007, 51.
- Granger, D.N.; Kvietys, P.R. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol. 2015, 6, 524–551.
- Panigrahi, M.; Sadguna, Y.; Shivakumar, B.R.; Kolluri, S.V.R.; Roy, S.; Packer, L.; Ravindranath, V. α-Lipoic Acid Protects against Reperfusion Injury Following Cerebral Ischemia in Rats. Brain Res. 1996, 717.
- Shivakumar, B.R.; Kolluri, S.V.R.; Ravindranath, V. Glutathione Homeostasis in Brain during Reperfusion Following Bilateral Carotid Artery Occlusion in the Rat. Mol. Cell. Biochem. 1992, 111, 125–129.