Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Multiple myeloma (MM) is a malignancy of plasma cells which is rising in incidence in the developed world. Plasma cells are mature antibody-producing B cells which reside in the bone marrow and are essential for maintaining humoral immunity. Multiple myeloma is characterized by a monoclonal proliferation of plasma cells resulting in the production of monoclonal antibody and end-organ damage.
- multiple myeloma
- epidemiology
- etiology
- risk factors
1. Diagnosis
Patients often present with CRAB symptoms. When MM is suspected, blood and urine electrophoresis should be performed to look for the monoclonal light-chain secreted by the neoplasm. Blood levels of IgG, IgM, and IgA can identify the isoform of light-chain produced. If levels are elevated but under 3 g/dL, the disease can be classified as SM or MGUS instead of MM. A serum-free light-chain (FLC) assay is more sensitive than urine (where the light-chain protein is called a Bence–Jones protein). Serum albumin and beta-2 microglobulin (B2M) from bloodwork are also valuable for diagnosing and staging the disease. The definitive diagnosis requires a bone marrow biopsy with greater than 10% clonal plasma cells, or the presence of a plasmacytoma elsewhere. End-organ damage is necessary to distinguish from SM. Renal indices should be evaluated to a glomerular filtration rate, usually calculated by creatinine levels, that can be used to establish renal insufficiency. Imaging, such as CT (without contrast dye due to renal damage), MRI, and PET scans, is used to uncover lytic bone lesions. If patients are unable to undergo these imaging procedures, a skeletal survey is used instead. Eighty percent of patients have some skeletal lesions, fractures, or osteopenia at the time of diagnosis [3,18,19]. The revised diagnostic criteria by the International Myeloma Working Group Diagnostic Criteria for MM and related Plasma Cell disorders are shown in Table 1 [20].
Table 1. The revised diagnostic criteria by the International Myeloma Working Group Diagnostic Criteria for MM and related Plasma Cell disorders [20]. CRAB: Hypercalcemia, Renal failure, Anemia, Bone pain; FLC: free light chains; MGUS: monoclonal gammopathy of undetermined significance.
| Disorder | Diagnostic Criteria |
|---|---|
| SMOLDERING MULTIPLE MYELOMA | Two criteria must be met:
|
| MULTIPLE MYELOMA | Two criteria must be met:
|
| IgM MGUS | Three criteria must be met:
|
| LIGHT-CHAIN MGUS | All the following criteria must be met:
|
2. Staging
The two main staging systems used for MM are the international staging system (ISS) and the Durie–Salmon system (DSS). The ISS stratifies cases into three stages: Stage I for those with a B2M less than 3.5 mg/L and albumin > 3.5 g/dL, Stage III for those with a B2M greater than 5.5 mg/L, and Stage II for all those in between [19]. The revised ISS adds prognostic information such as lactate dehydrogenase (LDH) levels and chromosomal abnormalities detected via FISH. In an international clinical trial with 3060 participants, for those with stage I disease, overall survival (OS) and progression-free survival (PFS) at five years was 82% and 55%, respectively. Median PFS was 66 months, and OS was not reached. For Stage II disease, these values were 62% and 36%, 42 months and 83 months, respectively. For Stage III disease, these values were 40%, 24%—29 months and 43 months, respectively. Further risk stratification can be made based on chromosomal translocation, many of which involve the IgH locus on chromosome 14 (14q32) [20].
With the advent of new therapies, the overall survival of the disease has increased. With improving survival, there was a need for re-staging the disease for early diagnosis, treatment, and preventing end-organ damage [20]. Until recently, MM was defined by the presence of a clonal process that correlates with end-organ involvement (presence of CRAB features). In patients with the absence of CRAB criteria, three biomarkers were included in the diagnostic criteria by the International Myeloma Working Group (IMWG) in 2014 [20]. In addition, the recommendations were to include CT and PET-CT to diagnose the involvement of bones. A new staging system has been developed that incorporates high-risk cytogenetic abnormalities in addition to standard laboratory markers of prognosis [20].
Both the ISS and DSS systems assess the tumor burden, but neither ISS nor DSS takes into consideration the biology of the disease, which determines the overall survival [20,21,22].
The revised international staging system (RISS) combines elements of tumor burden (ISS) and disease biology [23]. It was developed based on a study of 11 international trials. The 5 year survival rates among the patients with Stage I, II, and III RISS were 82%, 62%, and 40%, respectively [20].
3. Management
The survival has more than doubled over the past decades due to the introduction of new chemotherapy combinations, targeted small molecule inhibitors, and monoclonal antibodies. In Figure 7, we present a modified flow chart depicting the general outline of treatment options for MM adopted from mSMART.org. The approval of multiple active agents in the treatment of MM has generated numerous possible drug combinations that can be used in first-line and relapsed settings. The initial therapy of patients with symptomatic MM depends on risk stratification of the MM, the patient’s eligibility for autologous hematopoietic stem cell transplantation (HCT), and an assessment of the patient’s pre-existing comorbidities (e.g., presence of neuropathy or renal failure). For those ineligibles for transplant, a triplet regimen is also recommended and then followed with maintenance therapy until toxicity limits the use. One of the most commonly used front-line triple therapies is bortezomib (a proteasome inhibitor), lenalidomide (an immunomodulator that downregulates inflammatory and proliferative cytokines), and dexamethasone (a long-acting steroid), (called VRd after the tradenames Velcade, Revlimid, and Dexamethasone, respectively) [18,24].
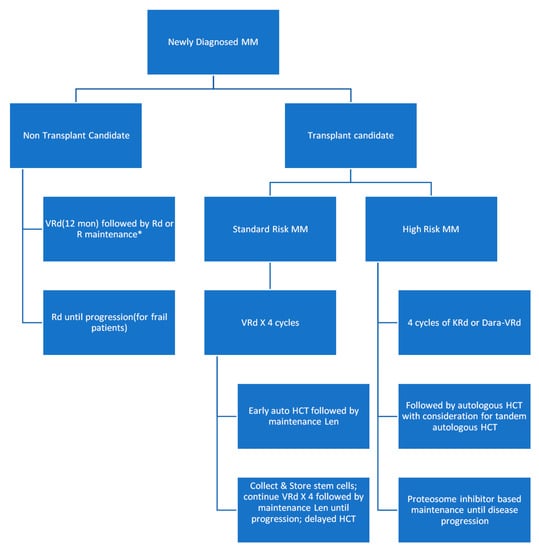
Figure 7. Modified flow chart depicting the general outline of treatment options for MM adopted from mSMART.org. HCT: hematopoietic stem cell transplantation.
The results of the SWOG S0777 trial showed that the triplet regimen VRd (i.e., bortezomib, lenalidomide, dexamethasone) improved response rates, depth of response, progression-free survival, and overall survival compared with the then approved front-line regimen Rd (lenalidomide, dexamethasone) making a three-drug regimen the main stay of initial therapy for most patients with MM [25]. This induction is typically followed by lenalidomide and dexamethasone maintenance. As of 2016, lenalidomide and bortezomib have been approved in 73 and 106 countries worldwide, respectively.
For most patients with standard-risk MM, the most commonly used induction therapy is VRd. VCd also called CyBorD (bortezomib, cyclophosphamide, dexamethasone) is an acceptable alternative for patients who are taught to have an increased risk of complication from the use of lenalidomide (e.g., patients with acute renal failure or those with increased thromboembolic risk) [26]. Other triplets like DRd (daratumumab, lenalidomide, and dexamethasone) and KRd (carfilzomib, lenalidomide, and dexamethasone) are also reasonable options for patients who cannot tolerate bortezomib (e.g., due to the fact of severe neuropathy) and for those who are not transplant candidates. Although OS data are immature, DRd has shown improved PFS when compared with Rd [27]. For frail patients who may not tolerate the increased toxicity that comes with triple regimens, Rd is still an acceptable alternative.
If patients relapse within 12 months of completion of initial therapy, this is a considered “high-risk” relapsed disease. Patients with a high risk of MM do poorly with conventional treatment options and are therefore encouraged to participate in clinical trials that employ novel therapeutic strategies. Outside of a clinical trial, VRd remains the preferred choice of initial chemotherapy for patients with high-risk MM who are not candidates for HCT [28,29]. KRd instead of VRd can be used as initial therapy in patients with t(4;14), t(14;16), t(14;20), and those with ≥2 high-risk abnormalities based on the results of phase 2 studies suggesting higher complete response (CR) rates and minimal residual disease negativity rates compared with historical results using VRd [28,30,31]. Albeit weak, there is data suggesting that use of a proteasome inhibitor may abrogate the prognostic impact of at least some of these high-risk genetic markers [32].
On the front line, autologous HCT is preferred for those who can tolerate it. Those undergoing HCT typically receive a triplet regimen for 3–4 months prior to stem cell collection in order to reduce tumor cell numbers in the bone marrow. Those who had a durable benefit from HCT in front-line or did not receive HCT in front-line but are now eligible are recommended an HCT in second line following induction [33,34]. Collected cells can be transplanted early or on a delayed schedule (after 8–12 cycles of the initial therapy or after first relapse). Allogeneic transplant remains under investigation for MM.
The recently approved monoclonal antibody, daratumumab, which targets CD38 (a receptor highly expressed on myeloma cells), has become a favorite for later line induction regimens [35,36]. Other antibodies approved include elotuzumab for the SLAMF7 receptor and isatuximab, which also targets CD38 (but comes with a greater risk of adverse events since it is chimeric). In addition, also used in relapsed myeloma are next generation proteasome inhibitors such as carfilzomib and ixazomib. Multiple myeloma relies on proteasomes to eliminate protein waste caused by ceaseless monoclonal light-chain production, thus inhibiting proteasomes selectively targets myeloma cells. Lastly, immunomodulators like lenalidomide, pomalidomide, and thalidomide are frequently used in triplet or quadruplet induction regimens and maintenance regimens [33,34].
Extramedullary disease can be seen in 5% of MM patients [37,38]. It can manifest in the skin, lymph nodes, abdomen, upper respiratory tract, and the central nervous system (CNS) [39].
Despite significant advances in the management of myeloma, the treatment for relapsed/refractory multiple myeloma (RRMM) continues to be challenging. Venetoclax, an inhibitor of the anti-apoptotic protein BCL-2, is very effective among patients with t(11;14). A review by Basali et al. [40] has shown an overall response of approximately 78% during their experience with venetoclax-based regimens in ten RRMM patients who had a median of six prior lines of therapy. Central nervous system involvement is not only a rare complication but also has a very poor prognosis. Recently, Egan et al. [41] provided a review of the current literature including the presentation, treatment, and survival data among patients with CNS involvement.
Other treatment considerations, including bisphosphonates, such as zoledronic acid, or bone-stimulating agents (BSAs), like denosumab, are recommended upon diagnosis in order to prevent or palliate lytic lesions and fractures [42,43]. Moreover, there is the need to use antiviral prophylaxis with acyclovir 400 mg BID throughout treatment with proteasome inhibitors, administration of bortezomib subcutaneously rather than IV, and once weekly rather than twice weekly to dramatically decrease the risk of peripheral neuropathy without compromising efficacy [44,45,46,47]. When using regimens that include lenalidomide (such as VRd, Rd, and KRd), consideration for the need of routine antithrombotic prophylaxis, dose adjustment for patients with creatinine clearance < 60, and recognition of the myelotoxic nature of lenalidomide to the bone marrow making peripheral stem cell collection difficult is very important. Low-dose dexamethasone (i.e., once weekly 40 mg dose) is associated with less dexamethasone-related toxicity than more intensive dosing scheme without compromising efficacy [48].
Several studies are underway to evaluate the effect of adding a fourth drug (such as daratumumab) to a triplet backbone but are not yet standard of care. Chimeric antigen receptor T cell therapy for the MM receptor BCMA is currently being investigated and has shown promise for refractory disease [49].
This entry is adapted from the peer-reviewed paper 10.3390/medsci9010003
This entry is offline, you can click here to edit this entry!