Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Orthopedics
椎间盘变性(IDD)是一种进行性和多因素的病理过程,主要与腰痛和永久性残疾有关。焦亡是一种由炎症小体和半胱天冬酶激活引发的裂解程序性细胞死亡。与细胞凋亡不同,焦亡的特征在于质膜破裂和炎症介质的释放,加速细胞外基质(ECM)的破坏。研究表明,在 IDD 的进展中,含有 pyrin 结构域的 3 (NLRP3) 炎症小体介导的髓核 (NP) 细胞焦亡被激活。
- pyroptosis
- intervertebral disc degeneration
- NLRP3 inflammasome
- nucleus pulposus
1. Pyroptosis Triggers Cell Death in IDD
Cell death, especially NP cell death, is one of the most critical factors triggering intervertebral disc (IVD) degeneration (IDD). It leads to a loss of cell function and a decline in nutrient synthesis and repair capability and, moreover, increases inflammation and oxidative stress [7]. All of this aggravates the degenerative cascade in IVDs. A positive correlation between NLRP3 expression and the degenerative score was found in 45 clinical samples [61]. This result demonstrated that pyroptosis is widely involved in IDD progression, which is mainly induced by NLRP3 inflammasome (Figure 1).
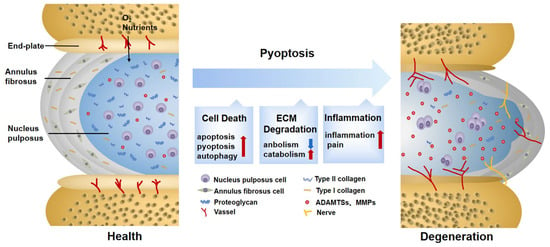
Figure 1. Changes in normal and degenerative intervertebral discs. The intervertebral disc is composed of nucleus pulposus (NP), annulus fibrosus (AF), and endplate (EP), together constituting a closed buffer system against stress. While in IDD, cell death, ECM degradation, and secondary verification aggravate the vicious cycle, which is closely associated with pyroptosis. ECM: extracellular matrix; ADAMTS: a disintegrin and metalloproteinase with thrombospondin motifs; MMPs: matrix metalloproteinases.
Besides directly causing cell death, pyroptosis and other programmed-cell-death pathways are jointly cross-regulated in the complex and harsh degenerative environment. It was found that, after lumbar spine instability surgery, the height of the IVD was reduced in mice, and fissures and folds were formed between the AF layers, with osteophyte formation in the EP. The TUNEL assay and pyroptosis-related protein detection showed that both apoptosis and pyroptosis in the IDD-model group were potentiated [62]. Studies on anti-inflammatory protein A20 further revealed that when pyroptosis was inhibited, the expression of inflammatory cytokines coupled with apoptosis was also markedly reduced [63]. The above results suggest that there is an interplay between apoptosis and pyroptosis in IDD. The molecular mechanism behind the interplay may be explained by PANoptosis [64], which is characterized by pyroptosis, apoptosis, and necroptosis. Li et al. [65] first discovered PANoptosis, consisting of pyroptosis and apoptosis, in IDD lesions. They used different concentrations of TNF-α to treat NP cells and found that both pyroptosis and apoptosis were upregulated. However, pyroptosis was predominant at low concentrations, whereas apoptosis was predominant at high concentrations, implying that the major type of cell death may vary during different periods of IDD progression [65]. This interconverting relationship may be partially explained by the altered concentrations of inflammatory mediators in the IDD microenvironment and the activation of the CASP3-GSDME pathway in the crosstalk of pyroptosis and apoptosis [66]. Moreover, the relationship between pyroptosis and autophagy is also intriguing. In bromodomain-containing protein 4-inhibited rat NP cells, enhanced autophagy was able to reduce pyroptosis to relieve degenerative development. Whether autophagy enhancement is linked with attenuated pyroptosis requires further investigation; however, it has been demonstrated that the NF-κB signaling pathway is involved [67]. Similarly, one study found a tendency for autophagic vacuoles to increase with higher microtubule-associated protein 1 light chain 3 (LC3)-II/LC3-I ratios in NLRP3 knockout NP cells, demonstrating that pyroptosis and autophagy are antagonistic to each other [68]. Moreover, some researchers have tried to explore the mechanism underlying this phenomenon. One study pointed out that autophagy protects against LPS-induced NP cell pyroptosis. The explicit regulatory relationship between them depends on SQSTM1/p62, one of the autophagy-related proteins, co-localizing with GSDMD-N. It degraded GSDMD-N through the autophagy–lysosome pathway, thereby attenuating pyroptosis [69]. In conclusion, the interplay between pyroptosis and other programmed cell deaths of NP cells in IDD currently remains questionable. It is suggested that subsequent experiments focus on this aspect, which may have a significant role in revealing the progression of IDD.
2. Pyroptosis Provokes ECM Disorder in IDD
ECM的组成、合成和重建对于IVD内部环境的稳定性极为重要。当ECM代谢受到严重干扰时,IVD的水合能力下降,变得更僵硬和脆弱,从而影响生物学功能[70]。先前的观察表明,焦亡可以诱导ECM降解。例如,烟酰胺磷酸核糖转移酶的下调通过NF-κB和MAPK途径阻断NLRP3的启动阶段,从而减少聚集聚糖和胶原蛋白II的降解[71]。胆固醇在IDD中积聚,显示聚集聚糖和胶原蛋白II的抑制,以及MMP13和ADTAMTS5的升高。这与NLRP3的激活期ER应激的激活有关[72]。尽管它们的信号不同,但它们都是DAMP,不仅可以激活NP细胞中的焦亡,还可以减少ECM合成并增加ECM降解。除了通过引起NP细胞死亡直接破坏ECM合成和分解的平衡外,焦亡释放的炎症介质和细胞内容物也在很大程度上参与ECM紊乱。NP细胞有助于ECM代谢[73]。当健康的NP细胞与焦解细胞共培养时,MMP13、ADAMTS4和ADAMTS5的表达上调[22]。它们水解的片段产物会破坏ECM的抗应激能力,并引发继发性炎症反应[74]。TNF-α和IL-1β是公认的驱动MMP和ADAMTSs合成和释放的因素[75],它们也与焦亡密切相关。上述介质可能导致焦亡引起的ECM降解。此外,这种现象也见于Modic EP改变和退行性AF组织,因此不仅限于NP细胞[61,62]。
此外,ECM紊乱也会加重焦亡。先前的研究发现,退化NP细胞中的厌氧糖酵解会产生酸性环境,激活ROS/NLRP3/半胱天冬酶-1介导的焦亡[23]。然而,一项研究得出了相反的结论。随着酸化加剧,外源性因素可能加剧ECM降解,而不是NP细胞NLRP3降低[76]。另一方面,代谢物(如晚期糖基化终产物)的积累使胶原纤维变硬并促进NLRP3的组装[77]。这在通过抑制焦亡来恢复ECM紊乱和延缓IDD进展方面显示出希望。注射半胱天冬酶-1抑制剂VX-76可防止纤维组织替代蛋白聚糖,有效延缓IVD的衰老和纤维化[69]。
3. 焦亡诱发IDD继发性炎症
继发性炎症细胞因子增加被认为是症状性IDD的重要特征[78]。IVD的内部区域缺乏血管化结构,并与宿主免疫系统隔离。NP细胞通常缺乏免疫耐受性,但能够吞噬细胞和分泌炎症因子。当暴露于炎症环境时,它们会产生固体自身免疫性和炎症级联反应[79,80]。IDD的炎症表现为三种方式,部分归因于焦亡。
IVD结构和生理学的改变导致炎症因子的产生,这是第一阶段最显著的特征[81]。焦亡的特征是典型的炎症细胞死亡模式,直接或间接地在IVD细胞中释放促炎因子[17]。作为焦亡的直接产物,IL-1β被认为是参与椎间盘性背痛的关键细胞因子[82]。同时,当暴露于IL-1β时,IL-6和IL-8等炎症因子的表达明显升高[83]。IDD中的某些物质可以通过诱导焦亡来引发炎症反应的情况并不少见。细胞外ATP(eATP)是来自ATP门控离子通道家族的NLRP3和P2X7R的代表共激活剂。当受到强烈刺激时,P2X7R转移到细胞质中与NLRP3共定位并释放转化生长因子β(TGF-β)和IL-1。它也与IVD炎症有关[84]。然而,eATP是否直接或通过焦亡引起炎症仍有待进一步阐述。虽然IDD现阶段主要以无菌性炎症为特征,但近年来感染因素逐渐引起人们的关注。其中,痤疮丙杆菌占IDD病因的13%-44%[85]。据报道,痤疮丙酸杆菌可通过ROS-NLRP3信号通路诱导NP细胞的焦亡。热解产物会加重IDD中炎症因子的聚集[22]。相反,注射NLRP3抑制剂MCC950成功改善了IDD的炎症环境[86],表明靶向焦亡可能是防止感染因素加重IDD炎症进展的治疗策略。然而,迄今为止,仍然缺乏对诱导焦亡的感染因子的特定抗原的深入见解。此外,尽管焦亡与IDD的炎症反应增强有关,但焦亡是否是IDD炎症的最初原因值得进一步讨论。
白细胞的浸润以及血管和神经的向内生长是第二阶段的事件。这个渐进过程的结果与第三阶段密切相关,因此将一起阐述。第三阶段由神经末梢致敏和伤害性神经元浸润引起的疼痛症状确定[81]。感觉神经纤维和血管通常分布在AF组织的外层,用于营养输送[87]。在小鼠腰椎-脊柱不稳定模型中,血管束和感觉神经在AF的内部和外部区域生长,降钙素基因相关肽(CGRP)阳性细胞增加[62]。这可能是由于焦亡释放的细胞因子会加剧ECM中蛋白聚糖的丢失并促进血管生成[88]。CGRP是控制炎症和疼痛的重要伤害性神经递质[89]。此外,细胞破裂释放的大量炎症因子不断刺激感觉神经,这也是一个显著的痛因。一项利用NF-κB抑制剂Bay11-7082对腰椎间盘突出症大鼠模型的研究发现,NLRP3的组装被阻断,CGRP和疼痛相关行为(如异常性疼痛和热痛阈值)得到改善。这表明NF-κB(疼痛和焦亡的标准信号)参与缓解神经性疼痛[90]。此外,虽然离子通道与炎症之间的关系尚未得到充分解释,但Ca2+-依赖性Pizol1通道通过机械拉伸[91]激活,ASIC3通道由酸感应[23]标记,可以促进焦亡,这可能会影响IVD后期疼痛的致敏[92]。
This entry is adapted from the peer-reviewed paper 10.3390/biom12121804
This entry is offline, you can click here to edit this entry!