Multiple system atrophy (MSA) is a rare neurodegenerative disease characterized by parkinsonism, cerebellar impairment, and autonomic failure. Although the causes of MSA onset and progression remain uncertain, its pathogenesis may involve oxidative stress via the generation of excess reactive oxygen species and/or destruction of the antioxidant system. One of the most powerful antioxidants is glutathione, which plays essential roles as an antioxidant enzyme cofactor, cysteine-storage molecule, major redox buffer, and neuromodulator, in addition to being a key antioxidant in the central nervous system. Glutathione levels are known to be reduced in neurodegenerative diseases. In addition, genes regulating redox states have been shown to be post-transcriptionally modified by microRNA (miRNA), one of the most important types of non-coding RNA. miRNAs have been reported to be dysregulated in several diseases, including MSA.
- multiple system atrophy
- neurodegenerative disease
- glutathione
- microRNA
- oxidative stress
- α-synuclein
1. Introduction
2. The Association between GSH Dysregulation and MSA
2.1. GSH Levels in Patients with MSA
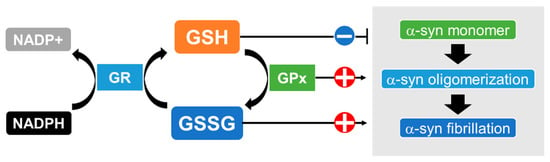
2.2. Possible Association of the Enzymes for GSH Synthesis and Metabolism with MSA
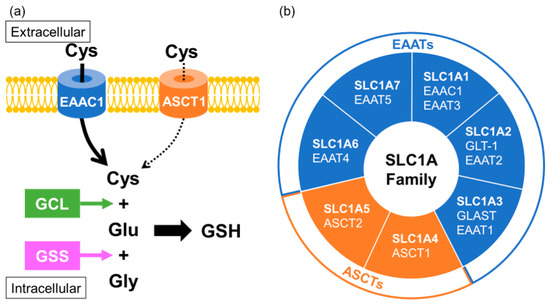
2.3. Association between Transporters Related to GSH Biosynthesis and MSA
3. The Association of miRNA Dysregulation and MSA Pathology
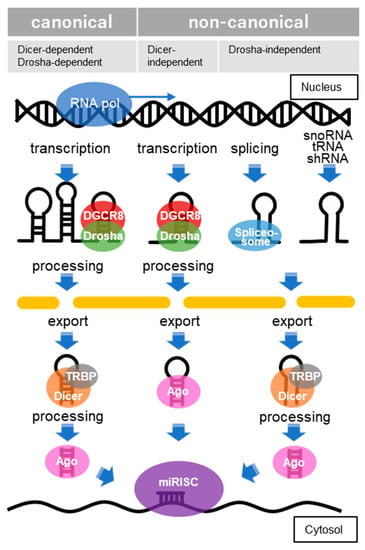
3.1. Molecular Mechanisms of miRNA Biogenesis
3.2. Dysregulation of Genes and miRNAs in Patients with MSA
3.3. Candidate miRNA Biomarkers for MSA
This entry is adapted from the peer-reviewed paper 10.3390/ijms232315076
References
- Fanciulli, A.; Wenning, G.K. Multiple-system atrophy. N. Engl. J. Med. 2015, 372, 249–263.
- Palma, J.A.; Norcliffe-Kaufmann, L.; Kaufmann, H. Diagnosis of multiple system atrophy. Auton. Neurosci. Basic Clin. 2018, 211, 15–25.
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469.
- Jellinger, K.A. Multiple System Atrophy: An Oligodendroglioneural Synucleinopathy1. J. Alzheimer’s Dis. JAD 2018, 62, 1141–1179.
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7.
- Mészáros, L.; Hoffmann, A.; Wihan, J.; Winkler, J. Current Symptomatic and Disease-Modifying Treatments in Multiple System Atrophy. Int. J. Mol. Sci. 2020, 21, 2775.
- Marmion, D.J.; Peelaerts, W.; Kordower, J.H. A historical review of multiple system atrophy with a critical appraisal of cellular and animal models. J. Neural Transm. 2021, 128, 1507–1527.
- Watanabe, H.; Saito, Y.; Terao, S.; Ando, T.; Kachi, T.; Mukai, E.; Aiba, I.; Abe, Y.; Tamakoshi, A.; Doyu, M.; et al. Progression and prognosis in multiple system atrophy: An analysis of 230 Japanese patients. Brain A J. Neurol. 2002, 125, 1070–1083.
- Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in Familial and Sporadic Multiple-System Atrophy. N. Engl. J. Med. 2013, 369, 233–244.
- Ogaki, K.; Fujioka, S.; Heckman, M.G.; Rayaprolu, S.; Soto-Ortolaza, A.I.; Labbé, C.; Walton, R.L.; Lorenzo-Betancor, O.; Wang, X.; Asmann, Y.; et al. Analysis of COQ2 gene in multiple system atrophy. Mol. Neurodegener. 2014, 9, 44.
- Quinzii, C.M.; Hirano, M.; DiMauro, S. Mutant COQ2 in multiple-system atrophy. N. Engl. J. Med. 2014, 371, 81–82.
- Lin, C.H.; Tan, E.K.; Yang, C.C.; Yi, Z.; Wu, R.M. COQ2 gene variants associate with cerebellar subtype of multiple system atrophy in Chinese. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 436–437.
- Zhao, Q.; Yang, X.; Tian, S.; An, R.; Zheng, J.; Xu, Y. Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: A case-control study and meta-analysis of the literature. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2016, 37, 423–430.
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, e2181–e2185.
- Danka Mohammed, C.P.; Park, J.S.; Nam, H.G.; Kim, K. MicroRNAs in brain aging. Mech. Ageing Dev. 2017, 168, 3–9.
- Kinoshita, C.; Aoyama, K.; Nakaki, T. microRNA as a new agent for regulating neuronal glutathione synthesis and metabolism. AIMS Mol. Sci. 2015, 2, 124–143.
- Kinoshita, C.; Kubota, N.; Aoyama, K. Interplay of RNA-Binding Proteins and microRNAs in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 5292.
- Ubhi, K.; Rockenstein, E.; Kragh, C.; Inglis, C.; Spencer, B.; Michael, S.; Mante, M.; Adame, A.; Galasko, D.; Masliah, E. Widespread microRNA dysregulation in multiple system atrophy—Disease-related alteration in miR-96. Eur. J. Neurosci. 2014, 39, 1026–1041.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Shukla, D.; Mandal, P.K.; Mishra, R.; Punjabi, K.; Dwivedi, D.; Tripathi, M.; Badhautia, V. Hippocampal Glutathione Depletion and pH Increment in Alzheimer’s Disease: An in vivo MRS Study. J. Alzheimer’s Dis. JAD 2021, 84, 1139–1152.
- Mandal, P.K.; Dwivedi, D.; Shukla, D.; Samkaria, A.; Roy, R.G.; Arora, Y.; Jindal, K. Interplay Between Hippocampal Glutathione Depletion and pH Increment in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2022, 88, 1–6.
- Chiang, G.C.; Mao, X.; Kang, G.; Chang, E.; Pandya, S.; Vallabhajosula, S.; Isaacson, R.; Ravdin, L.D.; Shungu, D.C. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol. 2017, 38, 1130–1137.
- Jenner, P. Presymptomatic detection of Parkinson’s disease. Journal of neural transmission. Supplementum 1993, 40, 23–36.
- Pearce, R.K.B.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677.
- Andronesi, O.C.; Nicholson, K.; Jafari-Khouzani, K.; Bogner, W.; Wang, J.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Schwarzschild, M.A.; et al. Imaging Neurochemistry and Brain Structure Tracks Clinical Decline and Mechanisms of ALS in Patients. Front. Neurol. 2020, 11, 590573.
- Chen, J.J.; Thiyagarajah, M.; Song, J.; Chen, C.; Herrmann, N.; Gallagher, D.; Rapoport, M.J.; Black, S.E.; Ramirez, J.; Andreazza, A.C.; et al. Altered central and blood glutathione in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Alzheimer’s Res. Ther. 2022, 14, 23.
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130.
- Fanciulli, A.; Stankovic, I.; Krismer, F.; Seppi, K.; Levin, J.; Wenning, G.K. Multiple system atrophy. Int. Rev. Neurobiol. 2019, 149, 137–192.
- Rink, C.; Khanna, S. Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid. Redox Signal. 2011, 14, 1889–1903.
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785.
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010.
- Shukla, G.S.; Hussain, T.; Srivastava, R.S.; Chandra, S.V. Glutathione peroxidase and catalase in liver, kidney, testis and brain regions of rats following cadmium exposure and subsequent withdrawal. Ind. Health 1989, 27, 59–69.
- Szymonik-Lesiuk, S.; Czechowska, G.; Stryjecka-Zimmer, M.; SŁomka, M.; MĄldro, A.; CeliŃski, K.; Wielosz, M. Catalase, superoxide dismutase, and glutathione peroxidase activities in various rat tissues after carbon tetrachloride intoxication. J. Hepato-Biliary-Pancreat. Surg. 2003, 10, 309–315.
- Dringen, R.; Hirrlinger, J. Glutathione Pathways in the Brain. Biol. Chem. 2003, 384, 505–516.
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free. Radic. Biol. Med. 2016, 95, 27–42.
- Fitzmaurice, P.S.; Ang, L.; Guttman, M.; Rajput, A.H.; Furukawa, Y.; Kish, S.J. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov. Disord. 2003, 18, 969–976.
- Jenner, P.; Dexter, D.T.; Sian, J.; Schapira, A.H.V.; Marsden, C.D. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental lewy body disease. Ann. Neurol. 1992, 32, S82–S87.
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355.
- Paik, S.R.; Lee, D.; Cho, H.-J.; Lee, E.-N.; Chang, C.-S. Oxidized glutathione stimulated the amyloid formation of α-synuclein. FEBS Lett. 2003, 537, 63–67.
- Xu, B.; Wu, S.-W.; Lu, C.-W.; Deng, Y.; Liu, W.; Wei, Y.-G.; Yang, T.-Y.; Xu, Z.-F. Oxidative stress involvement in manganese-induced alpha-synuclein oligomerization in organotypic brain slice cultures. Toxicology 2013, 305, 71–78.
- Tanaka, K.-I.; Sonoda, K.; Asanuma, M. Effect of Alteration of Glutathione Content on Cell Viability in α-Synuclein-Transfected SH-SY5Y Cells. Adv. Park. Dis. 2017, 6, 93.
- Clark, J.; Clore, E.L.; Zheng, K.; Adame, A.; Masliah, E.; Simon, D.K. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS ONE 2010, 5, e12333.
- Flohé, L. Glutathione peroxidase. Basic Life Sci. 1988, 49, 663–668.
- Kish, S.J.; Morito, C.L.; Hornykiewicz, O. Brain glutathione peroxidase in neurodegenerative disorders. Neurochem. Pathol. 1986, 4, 23–28.
- Koo, H.J.; Yang, J.E.; Park, J.H.; Lee, D.; Paik, S.R. α-Synuclein-mediated defense against oxidative stress via modulation of glutathione peroxidase. Biochim. Et Biophys. Acta 2013, 1834, 972–976.
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88.
- Probst-Cousin, S.; Bergmann, M.; Kuchelmeister, K.; Schröder, R.; Schmid, K.W. Ubiquitin-positive inclusions in different types of multiple system atrophy: Distribution and specificity. Pathol. Res. Pract. 1996, 192, 453–461.
- Aoyama, K.; Nakaki, T. Impaired glutathione synthesis in neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044.
- Janáky, R.; Varga, V.; Hermann, A.; Saransaari, P.; Oja, S.S. Mechanisms of L-cysteine neurotoxicity. Neurochem. Res. 2000, 25, 1397–1405.
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524.
- Hughes, C.E.; Coody, T.K.; Jeong, M.Y.; Berg, J.A.; Winge, D.R.; Hughes, A.L. Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 2020, 180, 296–310.e218.
- Richman, P.G.; Meister, A. Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J. Biol. Chem. 1975, 250, 1422–1426.
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59.
- Ferguson, G.; Bridge, W. Glutamate cysteine ligase and the age-related decline in cellular glutathione: The therapeutic potential of γ-glutamylcysteine. Arch. Biochem. Biophys. 2016, 593, 12–23.
- Njålsson, R. Glutathione synthetase deficiency. Cell. Mol. Life Sci. CMLS 2005, 62, 1938–1945.
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758.
- Divito, C.B.; Underhill, S.M. Excitatory amino acid transporters: Roles in glutamatergic neurotransmission. Neurochem. Int. 2014, 73, 172–180.
- Bianchi, M.G.; Bardelli, D.; Chiu, M.; Bussolati, O. Changes in the expression of the glutamate transporter EAAT3/EAAC1 in health and disease. Cell. Mol. Life Sci. 2014, 71, 2001–2015.
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126.
- Kanai, Y.; Hediger, M.A. The glutamate and neutral amino acid transporter family: Physiological and pharmacological implications. Eur. J. Pharmacol. 2003, 479, 237–247.
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine Transport through Excitatory Amino Acid Transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245.
- Sedláčková, L.; Laššuthová, P.; Štěrbová, K.; Vlčková, M.; Kudr, M.; Buksakowska, I.; Staněk, D.; Seeman, P. Severe neurodevelopmental disorder with intractable seizures due to a novel SLC1A4 homozygous variant. Eur. J. Med. Genet. 2021, 64, 104263.
- Abdelrahman, H.A.; Al-Shamsi, A.; John, A.; Ali, B.R.; Al-Gazali, L. A Novel SLC1A4 Mutation (p.Y191*) Causes Spastic Tetraplegia, Thin Corpus Callosum, and Progressive Microcephaly (SPATCCM) With Seizure Disorder. Child Neurol. Open 2019, 6, 2329048x19880647.
- Pironti, E.; Salpietro, V.; Cucinotta, F.; Granata, F.; Mormina, E.; Efthymiou, S.; Scuderi, C.; Gagliano, A.; Houlden, H.; Di Rosa, G. A novel SLC1A4 homozygous mutation causing congenital microcephaly, epileptic encephalopathy and spastic tetraparesis: A video-EEG and tractography—Case study. J. Neurogenet. 2018, 32, 316–321.
- Heimer, G.; Marek-Yagel, D.; Eyal, E.; Barel, O.; Oz Levi, D.; Hoffmann, C.; Ruzzo, E.K.; Ganelin-Cohen, E.; Lancet, D.; Pras, E.; et al. SLC1A4 mutations cause a novel disorder of intellectual disability, progressive microcephaly, spasticity and thin corpus callosum. Clin. Genet. 2015, 88, 327–335.
- Deng, X.; Sagata, N.; Takeuchi, N.; Tanaka, M.; Ninomiya, H.; Iwata, N.; Ozaki, N.; Shibata, H.; Fukumaki, Y. Association study of polymorphisms in the neutral amino acid transporter genes SLC1A4, SLC1A5 and the glycine transporter genes SLC6A5, SLC6A9 with schizophrenia. BMC Psychiatry 2008, 8, 58.
- Damseh, N.; Simonin, A.; Jalas, C.; Picoraro, J.A.; Shaag, A.; Cho, M.T.; Yaacov, B.; Neidich, J.; Al-Ashhab, M.; Juusola, J.; et al. Mutations in SLC1A4, encoding the brain serine transporter, are associated with developmental delay, microcephaly and hypomyelination. J. Med. Genet. 2015, 52, 541–547.
- Soma, H.; Yabe, I.; Takei, A.; Fujiki, N.; Yanagihara, T.; Sasaki, H. Associations between multiple system atrophy and polymorphisms of SLC1A4, SQSTM1, and EIF4EBP1 genes. Mov. Disord. Off. J. Mov. Disord. Soc. 2008, 23, 1161–1167.
- Kinoshita, C.; Aoyama, K. The Role of Non-Coding RNAs in the Neuroprotective Effects of Glutathione. Int. J. Mol. Sci. 2021, 22, 4245.
- Pérez-Soriano, A.; Bravo, P.; Soto, M.; Infante, J.; Fernández, M.; Valldeoriola, F.; Muñoz, E.; Compta, Y.; Tolosa, E.; Garrido, A.; et al. MicroRNA Deregulation in Blood Serum Identifies Multiple System Atrophy Altered Pathways. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 1873–1879.
- Kume, K.; Iwama, H.; Deguchi, K.; Ikeda, K.; Takata, T.; Kokudo, Y.; Kamada, M.; Fujikawa, K.; Hirose, K.; Masugata, H.; et al. Serum microRNA expression profiling in patients with multiple system atrophy. Mol. Med. Rep. 2018, 17, 852–860.
- Uwatoko, H.; Hama, Y.; Iwata, I.T.; Shirai, S.; Matsushima, M.; Yabe, I.; Utsumi, J.; Sasaki, H. Identification of plasma microRNA expression changes in multiple system atrophy and Parkinson’s disease. Mol. Brain 2019, 12, 49.
- Kim, T.; Valera, E.; Desplats, P. Alterations in Striatal microRNA-mRNA Networks Contribute to Neuroinflammation in Multiple System Atrophy. Mol. Neurobiol. 2019, 56, 7003–7021.
- Wakabayashi, K.; Mori, F.; Kakita, A.; Takahashi, H.; Tanaka, S.; Utsumi, J.; Sasaki, H. MicroRNA expression profiles of multiple system atrophy from formalin-fixed paraffin-embedded samples. Neurosci. Lett. 2016, 635, 117–122.
- Lee, S.-T.; Chu, K.; Jung, K.-H.; Ban, J.-J.; Im, W.-S.; Jo, H.-Y.; Park, J.-H.; Lim, J.-Y.; Shin, J.-W.; Moon, J.; et al. Altered Expression of miR-202 in Cerebellum of Multiple-System Atrophy. Mol. Neurobiol. 2015, 51, 180–186.
- Vallelunga, A.; Ragusa, M.; Di Mauro, S.; Iannitti, T.; Pilleri, M.; Biundo, R.; Weis, L.; Di Pietro, C.; De Iuliis, A.; Nicoletti, A.; et al. Identification of circulating microRNAs for the differential diagnosis of Parkinson’s disease and Multiple System Atrophy. Front. Cell. Neurosci. 2014, 8, 156.
- Starhof, C.; Hejl, A.M.; Heegaard, N.H.H.; Carlsen, A.L.; Burton, M.; Lilje, B.; Winge, K. The biomarker potential of cell-free microRNA from cerebrospinal fluid in Parkinsonian Syndromes. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 246–254.
- Seok, H.; Ham, J.; Jang, E.S.; Chi, S.W. MicroRNA Target Recognition: Insights from Transcriptome-Wide Non-Canonical Interactions. Mol. Cells 2016, 39, 375–381.
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610.
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485.
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16.
- Iwakawa, H.O.; Tomari, Y. Life of RISC: Formation, action, and degradation of RNA-induced silencing complex. Mol. Cell 2022, 82, 30–43.
- Stavast, C.J.; Erkeland, S.J. The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells 2019, 8, 1465.
- Kinoshita, C.; Aoyama, K.; Matsumura, N.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Rhythmic oscillations of the microRNA miR-96-5p play a neuroprotective role by indirectly regulating glutathione levels. Nat. Commun. 2014, 5, 3823.
- Kinoshita, C.; Kikuchi-Utsumi, K.; Aoyama, K.; Suzuki, R.; Okamoto, Y.; Matsumura, N.; Omata, D.; Maruyama, K.; Nakaki, T. Inhibition of miR-96-5p in the mouse brain increases glutathione levels by altering NOVA1 expression. Commun. Biol. 2021, 4, 182.
- Vallelunga, A.; Iannitti, T.; Capece, S.; Somma, G.; Russillo, M.C.; Foubert-Samier, A.; Laurens, B.; Sibon, I.; Meissner, W.G.; Barone, P.; et al. Serum miR-96-5P and miR-339-5P Are Potential Biomarkers for Multiple System Atrophy and Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 632891.
- Valera, E.; Spencer, B.; Mott, J.; Trejo, M.; Adame, A.; Mante, M.; Rockenstein, E.; Troncoso, J.C.; Beach, T.G.; Masliah, E.; et al. MicroRNA-101 Modulates Autophagy and Oligodendroglial Alpha-Synuclein Accumulation in Multiple System Atrophy. Front. Mol. Neurosci. 2017, 10, 329.
- Vidal-Martinez, G.; Segura-Ulate, I.; Yang, B.; Diaz-Pacheco, V.; Barragan, J.A.; De-Leon Esquivel, J.; Chaparro, S.A.; Vargas-Medrano, J.; Perez, R.G. FTY720-Mitoxy reduces synucleinopathy and neuroinflammation, restores behavior and mitochondria function, and increases GDNF expression in Multiple System Atrophy mouse models. Exp. Neurol. 2020, 325, 113120.
- Koepsell, H. General Overview of Organic Cation Transporters in Brain. In Organic Cation Transporters in the Central Nervous System; Daws, L.C., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 1–39.
- Baumgart, B.R.; Gray, K.L.; Woicke, J.; Bunch, R.T.; Sanderson, T.P.; Van Vleet, T.R. MicroRNA as biomarkers of mitochondrial toxicity. Toxicol. Appl. Pharmacol. 2016, 312, 26–33.
- Chang, L.; Xia, J. MicroRNA Regulatory Network Analysis Using miRNet 2.0. In Transcription Factor Regulatory Networks; Song, Q., Tao, Z., Eds.; Springer US: New York, NY, USA, 2022; pp. 185–204.
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017.
- Liu, L.; Liu, L.; Lu, Y.; Zhang, T.; Zhao, W. Serum aberrant expression of miR-24-3p and its diagnostic value in Alzheimer’s disease. Biomark. Med. 2021, 15, 1499–1507.
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic acids research 2019, 48, D127–D131.
- Yousefi, M.; Peymani, M.; Ghaedi, K.; Irani, S.; Etemadifar, M. Significant modulations of linc001128 and linc0938 with miR-24-3p and miR-30c-5p in Parkinson disease. Sci. Rep. 2022, 12, 2569.
- Di, G.; Yang, X.; Cheng, F.; Liu, H.; Xu, M. CEBPA-AS1 Knockdown Alleviates Oxygen-Glucose Deprivation/Reperfusion-Induced Neuron Cell Damage by the MicroRNA 24-3p/BOK Axis. Mol. Cell. Biol. 2021, 41, e0006521.
- Matoušková, P.; Hanousková, B.; Skálová, L. MicroRNAs as Potential Regulators of Glutathione Peroxidases Expression and Their Role in Obesity and Related Pathologies. Int. J. Mol. Sci. 2018, 19, 1199.
- Chis, A.R.; Moatar, A.I.; Dijmarescu, C.; Rosca, C.; Vorovenci, R.J.; Krabbendam, I.; Dolga, A.; Bejinar, C.; Marian, C.; Sirbu, I.O.; et al. Plasma hsa-mir-19b is a potential LevoDopa therapy marker. J. Cell. Mol. Med. 2021, 25, 8715–8724.
- Cao, X.Y.; Lu, J.M.; Zhao, Z.Q.; Li, M.C.; Lu, T.; An, X.S.; Xue, L.J. MicroRNA biomarkers of Parkinson’s disease in serum exosome-like microvesicles. Neurosci. Lett. 2017, 644, 94–99.
- Soto, M.; Iranzo, A.; Lahoz, S.; Fernández, M.; Serradell, M.; Gaig, C.; Melón, P.; Martí, M.J.; Santamaría, J.; Camps, J.; et al. Serum MicroRNAs Predict Isolated Rapid Eye Movement Sleep Behavior Disorder and Lewy Body Diseases. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 2086–2098.
- Joilin, G.; Gray, E.; Thompson, A.G.; Bobeva, Y.; Talbot, K.; Weishaupt, J.; Ludolph, A.; Malaspina, A.; Leigh, P.N.; Newbury, S.F.; et al. Identification of a potential non-coding RNA biomarker signature for amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa053.
- Arakawa, Y.; Itoh, S.; Fukazawa, Y.; Ishiguchi, H.; Kohmoto, J.; Hironishi, M.; Ito, H.; Kihira, T. Association between oxidative stress and microRNA expression pattern of ALS patients in the high-incidence area of the Kii Peninsula. Brain Res. 2020, 1746, 147035.
- Zhou, Y.; Wei, W.; Shen, J.; Lu, L.; Lu, T.; Wang, H.; Xue, X. Alisol A 24-acetate protects oxygen-glucose deprivation-induced brain microvascular endothelial cells against apoptosis through miR-92a-3p inhibition by targeting the B-cell lymphoma-2 gene. Pharm. Biol. 2021, 59, 513–524.
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780.